前言
在生信分析中,我们常常需要计算一个样本的几次实验结果或者不同样本实验结果的相关系数(样本间相关系数)以判断几个数据集之间相关的程度。
在本篇中及之后的内容中,为了用R得到相关系数热图(本篇中主要介绍了样本间的相关系数图,因为我刚好在做这个……),分别使用了pheatmap包(实验室的小伙伴推荐的)和corrplot包(我自己google找到的)进行了学习和实践,并对这些包中常用的参数进行了简单的介绍。
pheatmap包
pheatmap简介
官方介绍:
A function to draw clustered heatmaps whereone has better control over some graphical parameters such as cell size, etc
pheatmap实际上是 PrettyHeatmaps 的缩写,简单地来说,一个可以傻瓜式绘制聚类热图的R包。
常用参数介绍
基础设置
main 图的名字
file 要保存图的名字
color 表示颜色,赋值渐变颜色调色板colorRampPalette属性,选择“绿,黑,红”渐变,分为100个等级,,例:color = colorRampPalette(c(“navy”, “white”, “firebrick3”))(102)
sclae 表示值均一化的方向,或者按照行或列,或者没有,值可以是”row”, “column” 或者”none”
margins 表示页边空白的大小
fointsize 表示每一行的字体大小
聚类相关设置
cluster_cols 表示进行列的聚类,值可以是FALSE或TRUE
cluster_row 同上,是否进行行的聚类
treeheight_row 设置row方向的聚类树高
treeheight_col 设置col方向的聚类树高
clustering_distance_row 表示行距离度量的方法
clustering_distance_cols 同上,表示列距离度量的方法
clustering_method 表示聚类方法,值可以是hclust的任何一种,如”ward.D”,“single”,“complete”(默认), “average”, “mcquitty”, “median”,“centroid”, “ward.D2”
legend设置
legend TRUE或者FALSE,表示是否显示图例
legend_breaks 设置图例的断点,格式:vector
legend_labels legend_breaks对应的标签 例:legend_breaks = -1:4, legend_labels = c(“0”,“1e-4”, “1e-3”, “1e-2”,“1e-1”, “1”)
单元格设置
border_color 表示热图上单元格边框的颜色,如果不绘制边框,则使用NA
cellheight 表示每个单元格的高度
cellwidth 表示每个单元格的宽度
单元格中的数值显示:
display_numbers 表示是否将数值显示在热图的格子中,如果这是一个矩阵(与原始矩阵具有相同的尺寸),则显示矩阵的内容而不是原始值。
fontsize 表示热图中字体显示的大小
number_format 设置显示数值的格式,较常用的有”%.2f”(保留小数点后两位),”%.1e”(科学计数法显示,保留小数点后一位)
number_color 设置显示内容的颜色
热图分割设置
cutree_rows 基于层次聚类(使用cutree)划分行的簇数(如果未聚集行,则忽略参数)
cutree_cols 基于层次聚类(使用cutree)划分列的簇数
annotation相关设置
annotation_row 行的分组信息,需要使用相应的行名称来匹配数据和注释中的行,注意之后颜色设置会考虑离散值还是连续值,格式要求为数据框
annotation_col 同上,列的分组信息
annotation_colors 用于手动指定annotation_row和annotation_col track颜色的列表。
annotation_names_row boolean值,显示是否应绘制行注释track的名称。
annotation_names_col 同上,显示是否应绘制列注释track的名称。
使用
安装
install.packages(“pheatmap”) #安装pheatmap包 library(pheatmap) #加载pheatmap包 ?pheatmap #查看pheatmap包里面的详细介绍 ?pheatmap::pheatmap #查看pheatmap包里pheatmap函数的具体参数
绘制样本间相关系数图(简单使用)
(1)加载数据集:all_data
all_data为数据框格式,共包含9696996行,5列(5个样本),如下图所示。

colnames(all_data) <- c( 's1', 's2','s3', 's4','s5') #为数据框指定列名
(2)求样本间的相关系数
matrix <- cor (all_data[1:5]) #数据框格式可直接使用cor函数求相关系数
得到的matrix:

(3)绘制相关系数热图
pheatmap(matrix)
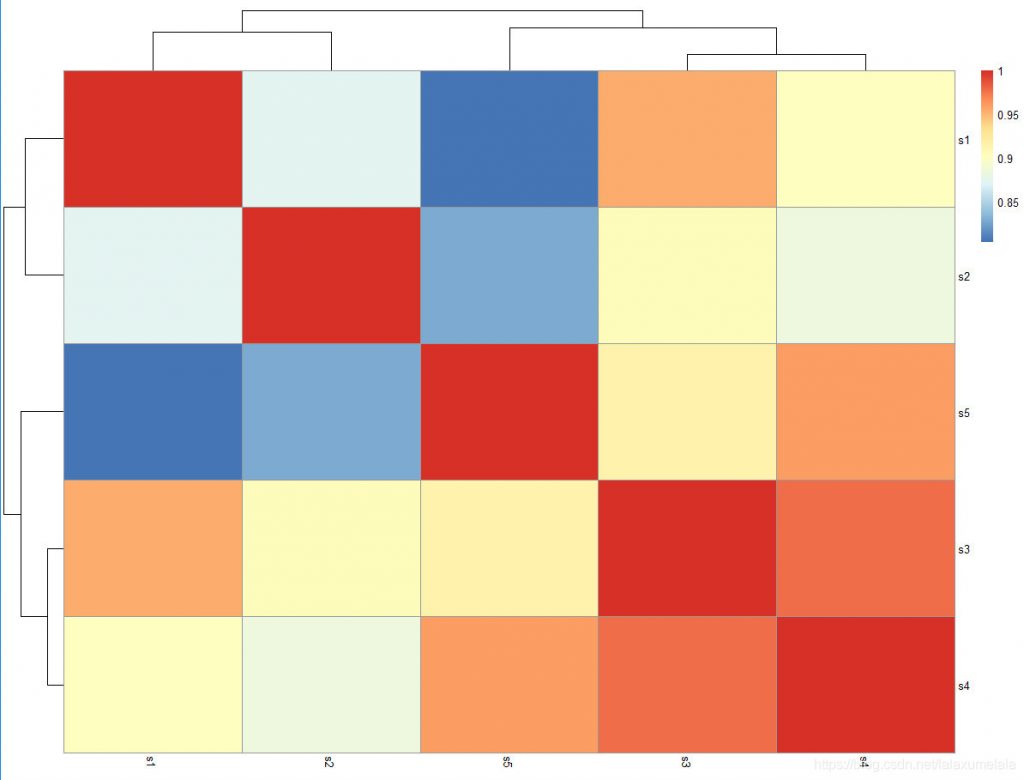
pheatmap(matrix,display_numbers=T) #在热图的单位格中显示数值

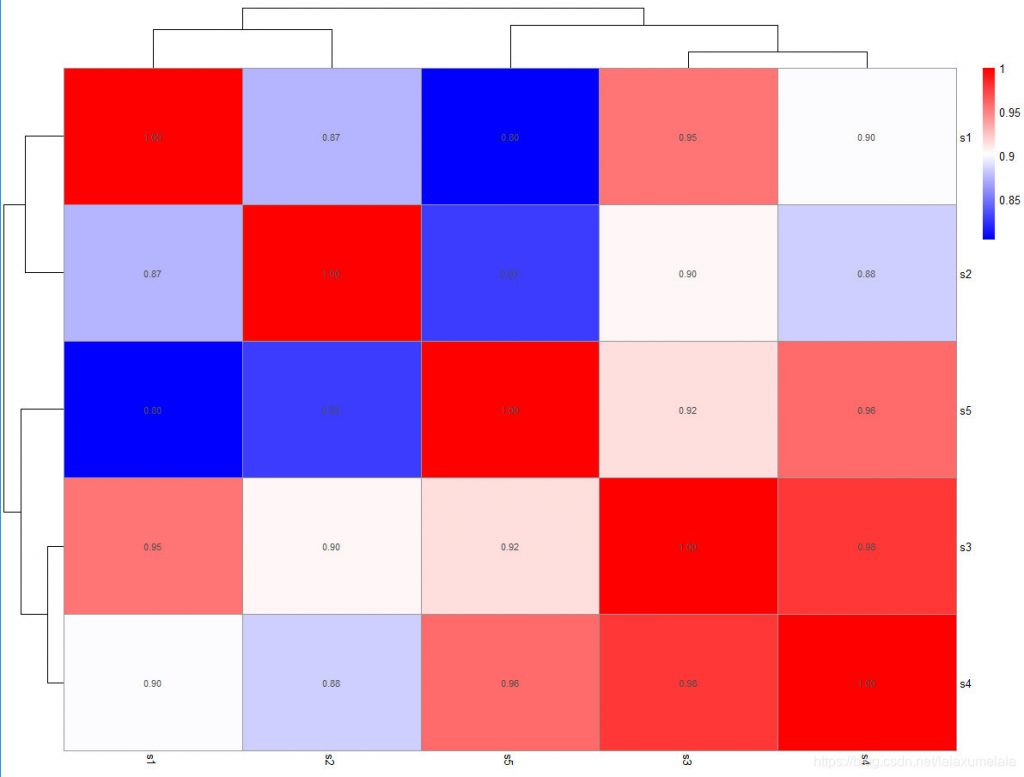
pheatmap(matrix,display_numbers=T,color=colorRampPalette(rev(c("red","white","blue")))(102)) #自定义颜色,使用红白蓝色系
pheatmap(matrix,display_numbers=T,fontsize=15) #fontsize设置热图中整体的字体为15

差异表达基因热图(进阶使用)
(1)生成测试数据集
#测试数据的生成参考了参考资料的第三个
test = matrix(rnorm(200), 20, 10)
test[1:10, seq(1, 10, 2)] = test[1:10,seq(1, 10, 2)] + 3
test[11:20, seq(2, 10, 2)] = test[11:20,seq(2, 10, 2)] + 2
test[15:20, seq(2, 10, 2)] = test[15:20,seq(2, 10, 2)] + 4
colnames(test) = paste("Test",1:10, sep = "")
rownames(test) = paste("Gene",1:20, sep = "")
(2)直接生成热图
pheatmap(test)

pheatmap(test,treeheight_row=100,treeheight_col=20) #设置col、row方向的树高

#取消列聚类,并且更改颜色
pheatmap(test,treeheight_row=100,treeheight_col=20,cluster_cols=FALSE,color=colorRampPalette(c("green","black","red"))(1000))

#取消单元格间的边框,调整字体大小,并且保存在桌面文件中
pheatmap(test,treeheight_row=100,treeheight_col=20,cluster_cols=FALSE,color=colorRampPalette(c("green","black","red"))(1000),border_color=NA,fontsize=10,fontsize_row=8,fontsize_col=16,file='C:/Users/xu/Desktop/test.jpg')

#增加分组信息,使得pheatmap显示行或列的分组信息
#这部分以及之后的内容参考了第四篇参考文献
annotation_col = data.frame(CellType =factor(rep(c("X1", "X2"), 5)), Time = 1:5) #增加Time,CellType分组信息
rownames(annotation_col) =paste("Test", 1:10, sep = "")
annotation_row = data.frame(GeneClass =factor(rep(c("P1", "P2", "P3"), c(10, 7,3)))) #增加GeneClass分组信息
rownames(annotation_row) =paste("Gene", 1:20, sep = "")
pheatmap(test, annotation_col =annotation_col, annotation_row = annotation_row)

#使用annotation_colors参数设定各个分组的颜色
ann_colors = list(Time =c("white", "green"),cellType = c(X1= "#1B9E77",X2 = "#D95F02"),GeneClass = c(P1 = "#7570B3", P2 ="#E7298A", P3 = "#66A61E"))
pheatmap(test, annotation_col =annotation_col, annotation_row = annotation_row, annotation_colors =ann_colors)

# cutree_rows,cutree_cols可以根据行列的聚类数将热图分隔开; pheatmap(test,cutree_rows=3,cutree_cols=2)

PS:当数值跨度比较大时,热图颜色变化会比较小,区分不开,可以使用以下命令:
bk = unique(c(seq(-2,2, length=100)))
pheatmap(test,breaks = bk)如何查看pheatmap的聚类结果
result <- pheatmap(test) summary(result)

# 行的聚类排列顺序 result$tree_row$order

# 得到行名的顺序 rownames(test)[result$tree_row$order]

# 查看按行聚类后的热图顺序结果 head(test[result$tree_row$order,])

# 查看按照行和列聚类之后,得到的热图顺序结果 head(test[result$tree_row$order,result$tree_col$order])

pheatmap总结
pheatmap总体来说使用起来比较简单,可以同时绘制热图和系统树图,参数的设置也很简单。此外,pheatmap包默认计算两两样本间的欧氏距离,然后利用欧式距离实现样本的聚类。