恶性实体瘤组织不仅包括肿瘤细胞,还包括与肿瘤相关的正常上皮和基质细胞,免疫细胞和血管细胞。基质细胞被认为在肿瘤生长、疾病进展和耐药性中起重要作用。
浸润性免疫细胞的作用与环境有关,虽然浸润性T淋巴细胞的抗肿瘤作用在卵巢癌中已被观察到,但在结直肠癌中,肿瘤的生长、侵袭和转移与肿瘤的生长、侵袭和转移有关.
对肿瘤组织中与肿瘤相关的正常细胞的全面了解可能为肿瘤生物学的研究提供重要的见解,并有助于开发可靠的预后和预测模型。
作者提出了一种新的算法,利用癌症样本转录谱的独特性质来推断肿瘤细胞的内容以及不同的浸润正常细胞,称为Estimation of STromal and Immune cells in MAlignant Tumour tissues using Expression data(ESTIMATE)。
作者重点研究基质细胞和免疫细胞,它们构成了肿瘤样本中主要的非肿瘤成分,并识别与肿瘤组织中基质细胞和免疫细胞浸润相关的特异性信号。通过进行单样本基因集富集分析(ssGSEA),作者通过计算基质和免疫评分来预测浸润基质和免疫细胞的水平,这些构成了在肿瘤组织中推断肿瘤纯度的 ESTIMATE score的基础。
ESTIMATE算法的概述如下图所示
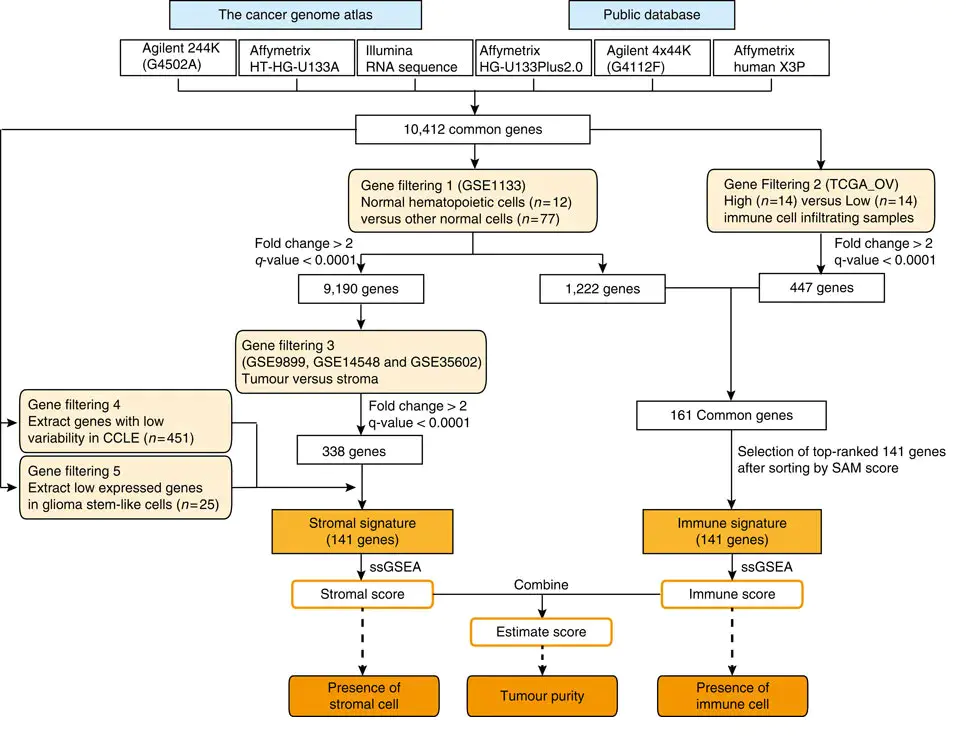
作者从不同平台的数据一筛选出两个 signature,一个是Stromal signature,Immune signature。两个标签分别有141个基因。通过ssGSEA分别计算基质得分和免疫得分。然后联合这两个得分来预测肿瘤纯度。
ESTIMATE适用平台有”affymetrix”, “agilent”, “illumina”,为了能让RNAseq数据能够适用ESTIMATE,使用VOOM法对RNAseq数据进行处理。此处用原始的sample文件进行输入
ESTIMATE代码
#安装包
if (F) {
library(utils)
rforge <- "http://r-forge.r-project.org"
install.packages("estimate", repos=rforge, dependencies=TRUE)
}
library(estimate)
help(package="estimate")
输入数据进行分析
rm(list = ls())
library(estimate)
in.file <- 'sample_input.txt' #输入文件
outfile2E <- 'ESTIMATE_input.gct' #生成ESTIMATE 的输入文件
outputGCT(in.file, outfile2E) #该函数以GCT格式写入输入文件
filterCommonGenes(input.f= in.file, output.f= outfile2E, id="GeneSymbol")
# 该功能将每个平台的不同数量的基因与10412个普通基因相结合。
### code chunk number 2: estimate
#这个功能计算基质,免疫,并估计得分每个样本使用基因表达数据。
estimateScore("ESTIMATE_input.gct", "ESTIMATE_score.gct")
plotPurity(scores="ESTIMATE_score.gct", samples="s516")
#根据ESTIMATE score绘制肿瘤纯度。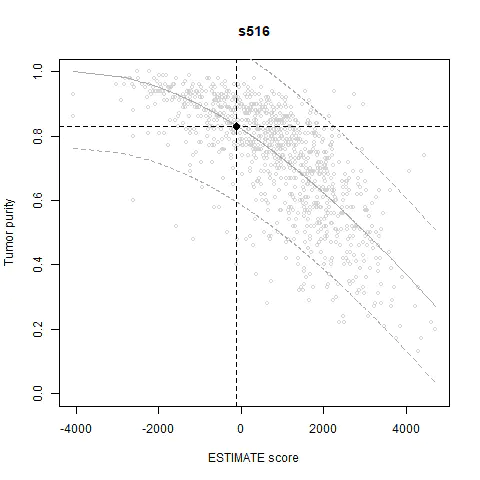
#将评分保存为txt格式
ESTIMATE_score <- read.table("ESTIMATE_score.gct", skip = 2,#前两行跳过
header = TRUE,row.names = 1)
ESTIMATE_score <- ESTIMATE_score[,2:ncol(ESTIMATE_score)]
ESTIMATE_score
write.table(ESTIMATE_score,file = "ESTIMATE_score.txt",quote = F,sep = "\t")
参考来源
Yoshihara, K., Shahmoradgoli, M., Martínez, E. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612 (2013).https://doi.org/10.1038/ncomms3612
您好,那么说,estimate是不是不应该应用于非肿瘤疾病的生信分析,可是我看到有文献使用它做免疫浸润评分来分组,这样可以吗
你好,请问我用TCGA的RNA-Seq数据输入 我的格式是counts fpkm 还是tpm呢
TPMs吧!后面还要log啊
您好,我是GEO数据库下载的芯片数据,与GPL进行了基因的匹配,匹配之后,得出的是航民为基因,列名为样本,都是GSE+数字的,我在哔站上看别人都是s+数字,我是不是数据处理方式不对呀。
您好 没有问题的 讲道理 GEO下载的芯片数据都有各自GSM***编号
您好请问免疫浸润输入文件只需要原发性肿瘤的样本对吗?别的和正常样本筛掉不要是吗?小白刚接触,可能问题有些傻?
没有关系的 什么问题都可以讨论
是的 如你所说 不需要正常样本