转自简书
使用R进行数据分析的时候,我们输入的数据大多为宽数据,为了适应R数据的作图要求,需要将宽数据转变为长数据。应该说这是长宽数据转变的重要原因之一。
什么是长数据
长数据一般是指数据集中的变量没有做明确的细分,即变量中至少有一个变量中的元素存在值严重重复循环的情况(可以归为几类),表格整体的形状为长方形,即 变量少而观察值多。
什么是宽数据
宽数据是指数据集对所有的变量进行了明确的细分,各变量的值不存在重复循环的情况也无法归类。数据总体的表现为 变量多而观察值少。
1. 为什么要进行数据长宽变换
变为长数据的一点原因就是上面提到的一点原因就是便于R 进行图形操作处理。而变为宽数据主要还是便于观察,以及数据清洗完成后,转变为宽数据,便于其他软件的操作和处理,例如excel,SPSS。
2. 宽数据变长数据
宽数据变长数据时,不会出现太多的问题,比较简单,使用tidyr package中的pivot函数即可顺利完成。好了,直接上例子。我们使用的例子是统计学方法选择(3)中survimer package 的骨髓瘤基因表达量数据集。
library(survminer)
data("myeloma")
head(myeloma)[1:3,1:11]
数据是这个样子

包括了分子分组,染色体的位置,治疗,与生存计算有关的数据,以及后面的几个基因所对应的表达量。先看看数据的数量
> dim(myeloma)
[1] 256 11
有256行,也就是256个观察单位,样本。
为了进一步研究,或者利用ggplot2作图,就要把后面的基因和表达量重新制定两列,变为长数据
library(tidyr)
library(dplyr)
library(tibble)
myeloma_longer<- myeloma %>%
rownames_to_column(var = 'sample') %>%
pivot_longer( cols = c("CCND1":"WHSC1"),
names_to = 'gene',
values_to = 'expr')
现在得到的数据是这样
> dim(myeloma_longer)
[1] 1536 8
> head(myeloma_longer)
# A tibble: 6 x 8
sample molecular_group chr1q21_status treatment event time gene expr
<chr> <fct> <fct> <fct> <int> <dbl> <chr> <dbl>
1 GSM50986 Cyclin D-1 3 copies TT2 0 69.2 CCND1 9908.
2 GSM50986 Cyclin D-1 3 copies TT2 0 69.2 CRIM1 421.
3 GSM50986 Cyclin D-1 3 copies TT2 0 69.2 DEPDC1 524.
4 GSM50986 Cyclin D-1 3 copies TT2 0 69.2 IRF4 16156.
5 GSM50986 Cyclin D-1 3 copies TT2 0 69.2 TP53 10
6 GSM50986 Cyclin D-1 3 copies TT2 0 69.2 WHSC1 262.
可以看到基因重复出现,与前面的样本信息进行循环匹配。
得到的数据就可以轻松作图了
p <- ggboxplot(myeloma_longer, x="gene", y="expr", color = "molecular_group", palette = "lancet", add = "jitter")
m1=p+stat_compare_means(aes(group=gene)) #默认是Wilcoxon
m1
按照上面的代码得到的图如下
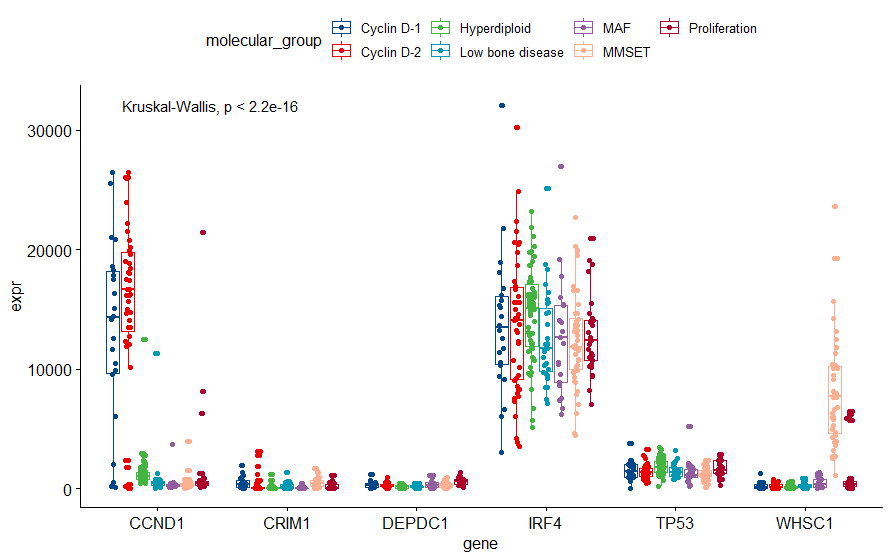
是以基因为横坐标分大组,然后又以分子标记分小组进行比较,可以观察各个组别之间的差异。
下面是另外的分组方法
p <- ggboxplot(myeloma_longer, x="molecular_group", y="expr", color = "gene", palette = "lancet", add = "jitter")
m1=p+stat_compare_means(aes(group=gene),label = "p.signif") #默认是Wilcoxon
m1
得到的图形如下
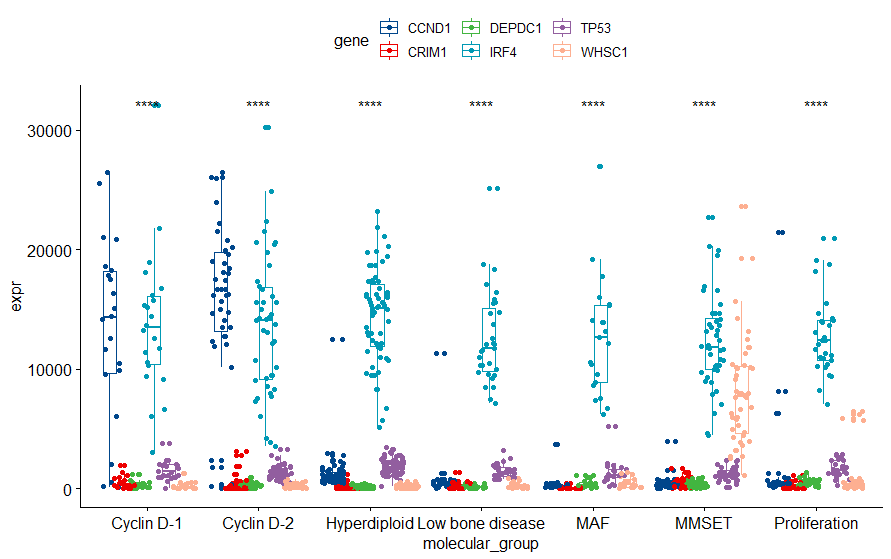
上面只是利用分子标志物进行了分组上的变换,发现每组差异性都很显著,其实从图上一看也很明显,结合其他分组,还可以又很多分组,年龄、治疗等等。图有了,就看怎么结合自己的研究课题,写点打动审稿人的故事了。
所以说,分组、画图、看差异、编故事,一般科研就这么出来了。
10.12小更新:有些时候横坐标的项目比较多,直接横行排列,显得臃肿,加上一句代码,让横坐标转换角度,这样就使得横坐标清新了然。
p <- ggboxplot(myeloma_longer, x="molecular_group", y="expr", color = "gene", palette = "lancet", add = "jitter")+
rotate_x_text(angle = 40)
3. 长数据变宽数据
利用tidyr packa进行长数据变换为宽数据时,会有一点小问题,主要是因为利用tidyr进行数据处理时,使用的都是tibble格式的数据模式,有别于data.frame,所以,在行的转换上需要注意。
3.1无差错转换
还是利用刚才转换成长数据的数据
是完全一致的,没有出现因为行名问题而导致的错误,主要是在转化为长数据的时候已经将样本名重新命名了一列。
经常会出现not uniquely identified的警告提示,不能完成长数据转变为宽数据。
3.2 出现错误提示
使用以下例子进行说明
data <- iris[, c(4,5)]
data %>% pivot_wider(names_from = Species,
values_from = Petal.Width)
出现错误提示
Warning message:
Values in `Petal.Width` are not uniquely identified; output will contain list-cols.
* Use `values_fn = list(Petal.Width = list)` to suppress this warning.
* Use `values_fn = list(Petal.Width = length)` to identify where the duplicates arise
* Use `values_fn = list(Petal.Width = summary_fun)` to summarise duplicates
解决方法:以name_from 为分组,给value一个标识
data <- iris[c(4,5)]
dat <- data %>%
group_by(Species) %>%
mutate(index = row_number()) %>%
pivot_wider(names_from = Species,
values_from = Petal.Width) %>%
select(-index)
dat
结果如下
> dat
# A tibble: 50 x 3
setosa versicolor virginica
<dbl> <dbl> <dbl>
1 0.2 1.4 2.5
2 0.2 1.5 1.9
3 0.2 1.5 2.1
4 0.2 1.3 1.8
5 0.2 1.5 2.2
6 0.4 1.3 2.1
7 0.3 1.6 1.7
8 0.2 1 1.8
9 0.2 1.3 1.8
10 0.1 1.4 2.5
# ... with 40 more rows
实际上就是给每一行给了一个单独的编号,这样就使的长数据变化为宽数据时,有了固定的位置,解决了错误提示中的Values in Petal.Width are not uniquely identified。
好了,长数据变为宽数据后,结合自己在统计学中所写的,就可以愉快地分组,比较差异,分析差异,写一个让审稿人信服的故事,然后发文了。当然想的美了,怎么分组,还是值得研究的问题。
参考书目及文章
https://blog.csdn.net/Ray_zhu/article/details/78679913
https://www.jianshu.com/p/e941f12179f3
https://blog.csdn.net/anna_datahummingbird/article/details/80348074