转载自:『珍藏版』Cancer Cell综述 | 通过靶向铁死亡消除肿瘤 – 知乎
肿瘤研究中的一个关键挑战是——在保持健康细胞完整的同时如何有效杀死肿瘤细胞。肿瘤细胞经常在死亡机制方面有缺陷,而这也是治疗失效的一个主要原因。为了满足生长需要,与普通的非肿瘤细胞相比,肿瘤细胞对铁的需求更高。这种对铁的依赖使得肿瘤细胞更容易发生铁催化的坏死,即铁死亡(最近Nature杂志背靠背发表两篇铁死亡的文章将该领域又向前推动了一大步,详见此前BioArt的报道:Nature背靠背 | 铁死亡调控新途径)。从FDA(食品药品管理局)许可的药物中鉴定铁死亡的诱导物,有希望通过诱发铁死亡杀死治疗失效的肿瘤,使得铁死亡成为治疗肿瘤的一个有前景的新方案。
2019年5月16日,来自比利时的Tom Vanden Berghe团队在Cancer Cell上发表了综述文章Targeting Ferroptosis to Iron Out Cancer。在这篇综述中,作者们首先简单描述了目前对铁死亡的诱导、执行、调节的分子机制的理解。接下来,详细描述了依赖诱导铁死亡的肿瘤治疗策略,并展望了这个新领域的未来。

前言
虽然医疗水平明显提高了,但根据世界卫生组织2018年的报告:肿瘤依然是全球第二大致死因素。用抗癌药触发凋亡性细胞死亡是杀死肿瘤细胞的主要途径。然而,由于肿瘤细胞对凋亡获得性的、内在的抵抗,在肿瘤中诱导凋亡的效果很有限。因此,利用其他形式的非凋亡性的细胞死亡来清除肿瘤细胞、控制抗药细胞克隆的扩增提供了新的治疗可能。二十余集中的细胞死亡研究发现存在多种调节性坏死的形式。程序性坏死和铁死亡作为两种形式的调节性坏死,正被作为备选方式用以清除抵抗凋亡的肿瘤细胞。越来越多的化合物和抗肿瘤药物被报道可以触发程序性坏死和铁死亡。程序性坏死作为研究详尽的调节性坏死形式,是由RIPK3和MLKL协同作用介导的。铁死亡是一种由铁催化、多不饱和脂肪酸的过度氧化介导的调节性坏死。最近,由于肿瘤中参与起始和执行程序性坏死的基因一般会下调或沉默,铁死亡获得了诸多关注。
铁死亡的实施:不饱和膜生锈了
铁死亡是由小分子erastin和RSL3诱发的一种独特的细胞死亡。起初,在过表达突变癌基因HRAS的基因工程细胞中,erastin和RSL3表现出了它们选择性的细胞毒性。然而,进一步的研究发现erastin对RAS突变的肿瘤细胞系没有选择性杀伤能力。
形态上,铁死亡的细胞表现出典型的坏死性形态,并伴随同质异形的小线粒体:嵴减少、膜凝聚、外膜破裂。此外,铁死亡细胞不展现任何凋亡的特征,敲低程序性坏死的调节分子RIPK1和RIPK3的细胞仍然能够发生铁死亡。铁死亡的实施是以由铁催化的含多不饱和脂肪酸的磷脂的过度氧化为特征的。含多不饱和脂肪酸的磷脂在哺乳动物细胞膜上大量存在。给细胞提供多不饱和脂肪酸会促进铁死亡,但是换成氘化的多不饱和脂肪酸就会使得细胞对氧化不敏感,敲除促进多不饱和脂肪酸吸附进磷脂的基因抑制铁死亡的发生。机制上,多不饱和脂肪酸中靠近亚甲基基团的双键削弱了双烯丙基中亚甲基基团的能量,导致脱氢和随后的氧化反应易于发生,而这些结果可以通过氧化脂质组学的方式检测到。铁死亡中脂质过氧化的毒性可以被铁螯合剂(如:去铁胺DFO)或多种亲脂原子团陷阱(维生素E, Fer-1,Lip1)中和。脂质的自发氧化
非酶催化的或自发的脂质的过氧化是一种自由原子团驱动的链式反应,其中,活性氧ROS起始了多不饱和脂肪酸的氧化。活性氧最典型的化学形式是羟基(OH-),羟基是氧自由基ROS高度灵活的水溶性的形式,可以起始脂质的过氧化。芬顿(Fenton)反应或芬顿样反应,主要是双氧水和过渡金属(如二价铁离子Fe2+)之间的反应,是羟基的主要来源。作为非酶催化的脂质过氧化反应的第一步,多不饱和脂肪酸的氢被羟基获取后形成以碳为中心的脂质原子团(L-)。氧分子与脂质原子团的迅速反应产生脂质过氧化原子团(LOO-)。随后,脂质过氧化原子团从临近的多不饱和脂肪酸获取氢形成氢过氧化物(LOOH)和一个新的脂质原子团,而这个新的脂质原子团可以起始另一次脂质原子团的链式反应。脂质氢过氧化物在存在二价铁离子的情况下又会转化成烷氧基原子团(LO-),烷氧基原子团随后与临近的多不饱和脂肪酸反应又可以起始另一次脂质原子团链式反应。
这个自发的过程是由铁和氧气催化的,当阻止脂质过氧化的分子监护机制失灵时,膜被毁掉、细胞死亡。作为主要的非酶催化的防御机制,链断裂的亲脂抗氧化剂可以通过提供电子中和脂质原子团。另外,当存在高浓度原子团时,两个原子团可以相互反应、形成稳定的非原子团分子。
酶催化的脂质氧化
酶催化的脂质过氧化反应是由一个脂氧合酶(LOX)家族的活性控制的。LOXs是一类非血红素的含铁的酶,可以催化自由的和酯化的多不饱和脂肪酸脱氧产生多种脂质氢过氧化物。在哺乳动物细胞中,亚麻油酸LA和花生四烯酸AA是LOXs最丰富的多不饱和脂肪酸底物。LOX5通过氧化花生四烯酸的第5位碳,合成5-过氧化氢二十碳四烯酸(5-HPETE)。LOX12和LOX15通过氧化花生四烯酸分别合成12-过氧化氢二十碳四烯酸(12-HPETE)和15-过氧化氢二十碳四烯酸(15-HPETE),通过氧化亚麻油酸分别合成9-过氧化氢十八碳二烯酸(9-HPODE)和13-HPODE。LOX5需要提前通过胞质磷脂酶A2(cPLA2)将酯化的花生四烯酸从膜磷脂中水解下来。而与LOX5不同,LOX12和LOX15可以直接氧化含有花生四烯酸的磷脂。抑制或敲低脂氧酶,在一些细胞系中抑制了铁死亡,这说明脂氧酶LOXs在铁死亡中扮演着重要的角色。近期有报道称维生素E除了它的亲脂原子团陷阱活性,也具有抑制LOX的活性,而这可以解释它对铁死亡潜在的抑制效果。
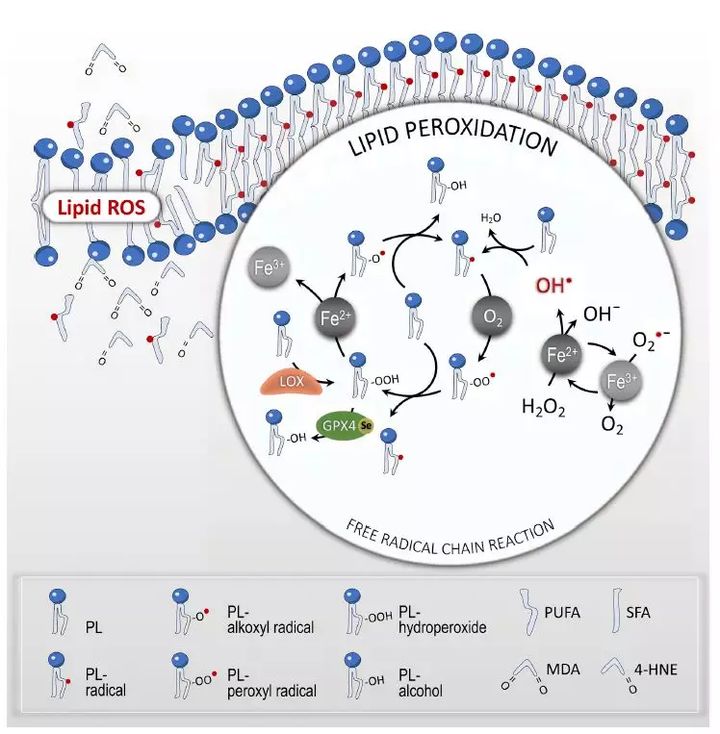
脂质氧化的毒性
氧化下游导致铁死亡的准确机制还有待解析。这些机制包括形成结构化的脂质孔,这种脂质孔类似在程序性坏死和细胞焦亡中观察到的蛋白质孔。此外,持续氧化或耗竭多不饱和脂肪酸可能会改变膜的流动性和结构,增加膜的通透性,最终导致膜完整性的丧失。人们使用分子动态模型推测:在铁死亡过程中,氧化剂使膜进入越来越稀疏、弯曲的恶性循环,最终破坏膜的稳定性,形成小孔和束状物。此外,脂质氢过氧化物可能会被分解成有毒的醛类,例如4-羟基-2-烯醛(4-HNEs)或丙二醛,它们交互作用可能使参与细胞内重要过程的蛋白失活,进而促进铁死亡。
经典的铁死亡的诱导
经典通路通过使膜抵抗过氧化损伤的主要保护性机制失活来诱导铁死亡。一种主要的膜保护性机制借助了谷胱甘肽过氧化物酶(GPXs)的活性。在GPXs家族分子中,GPX4可以抑制复杂脂质(如:磷脂和胆固醇)的氢过氧化反应,即使当磷脂和胆固醇被嵌入到膜和脂蛋白中时也可以发挥抑制作用。因此,GPX4被认为是保护生物膜不受氢过氧化反应的唯一GPX分子。通过耗竭细胞内的谷胱甘肽(GPX4必备的协同分子)等直接或间接靶向膜的机制,GPX4失活。
i) 细胞内谷胱甘肽的耗竭
Xc-系统由二硫键连接的异二聚体SLC7A11(xCT)和SLC3A2(4F2hc)组成,它可以输入细胞外的氧化形式的半胱氨酸,即胱氨酸,交换细胞内的谷氨酸。胱氨酸是谷胱甘肽合成所必需的,抑制胱氨酸的输入,可以最大程度地耗竭细胞内谷胱甘肽的水平。谷胱甘肽GSH是三肽形式的抗氧化剂,可以作为硒依赖性GPX4的协同分子,辅助GPX4减少脂质氢过氧化物。因此,通过erastin耗竭GSH间接灭活GPX4,导致有毒的脂质自由基ROS的积累,进而促进脂质过氧化。值得注意的是,1950年代和1970年代的早期观察发现剥夺胱氨酸可以抑制培养细胞的生长,这一类细胞死亡可以被亲脂抗氧化剂和铁螯合剂抑制。直接抑制谷胱甘肽的合成在一些细胞中足以诱导铁死亡。例如,在一些细胞中,用丁硫氨酸亚砜胺BSO抑制谷氨酸-半胱氨酸连接酶(GCL,一种参与原位合成GSH的酶)可以诱导铁死亡。
ii)GPX4的灭活或耗竭
通过化学蛋白质组学试验鉴定铁死亡诱导剂RSL3可以共价结合到GPX4位于活性位点的硒代半胱氨酸上,从而直接抑制了GPX4的磷脂过氧化酶活性。在细胞中过表达GPX4使得细胞抵抗RSL3诱导的铁死亡,而敲低GPX4促进铁死亡。其它化合物如ML162、withaferin A(WA)和食品药品管理局FDA批准的抗癌药altretamine也可以通过灭活GPX4诱导铁死亡。FIN56通过结合并激活参与胆固醇合成的鲨烯合酶促进铁死亡。这将导致对一些代谢物,如脂溶性抗氧化剂辅酶Q10(coQ10)和Sec-tRNA的抑制,导致耗竭或灭活GPX4。类似地,敲除GPX4会导致脂质氧自由基的迅速累积,而这些脂质氧自由基可以被亲脂原子团陷阱和铁螯合剂抑制。
非经典的铁死亡的诱导
在脂质过氧化机制中,很明显铁在铁死亡中扮演了重要的角色。铁池主要以二价铁离子Fe2+的形式存在(叫作不稳定铁池),可以通过Fenton反应直接催化自由原子团的形成,并且能进一步扩大脂质过氧化反应。此外,铁和铁的衍生物,如亚铁血红素或硫化铁Fe-S簇对于氧自由基生成酶(如NADPH、NOXs、LOXs和线粒体电子转运复合体)的活性是关键的。
非经典铁死亡的诱导指的是通过增加“不稳定铁池”而起始的铁死亡。比如:过度激活亚铁血红素加氧酶1(HMOX1)、减少铁转运蛋白(ferroportin)的表达或增加转铁蛋白(transferrin)的表达。添加全铁转铁蛋白(holo-transferrin)这种铁运输蛋白,也可以在Bax/Bak双敲除的细胞中剥夺氨基酸,解除铁死亡性的细胞死亡。虽然其中的机制仍不清楚,但是给细胞过量加载铁(如氯化铁、血红蛋白、氯高铁血红素、硫酸亚铁铵)可以充分诱导铁死亡。
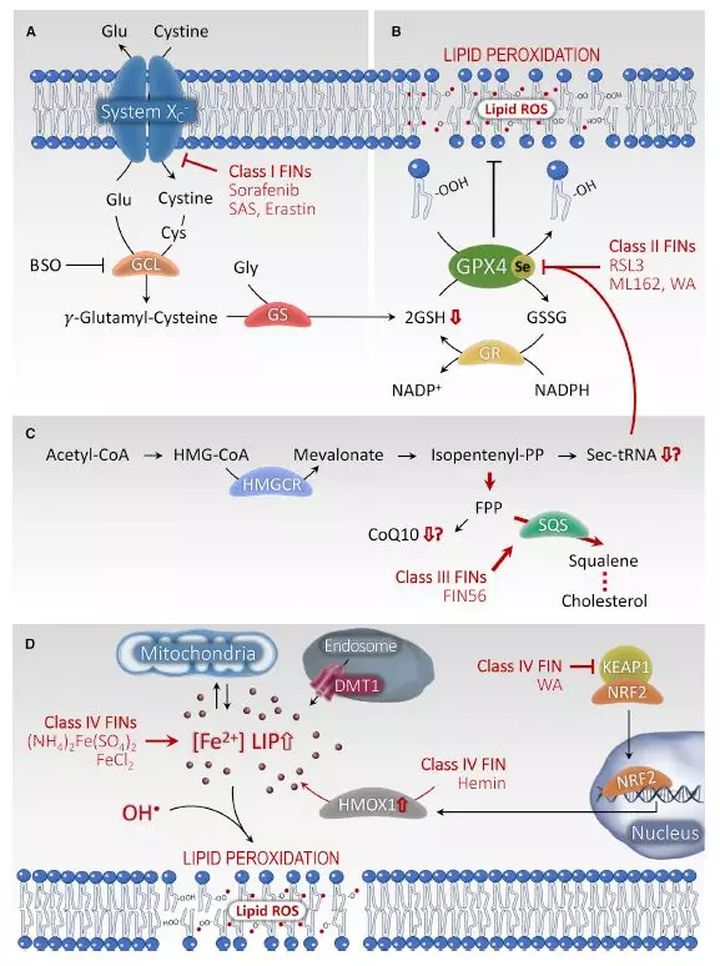
铁死亡的调节器
考虑到铁死亡是氧自由基、铁和多不饱和脂肪酸参与的代谢紊乱的结果,鉴定发现多种参与铁和能量代谢、脂质合成以及氧化应激的基因与通路具有调节铁死亡敏感性的潜力。
铁代谢
铁是一种氧化还原反应活跃的金属,它参与自由原子团的形成和脂质过氧化反应的扩大。因此,提高铁的水平可以使铁死亡易于发生。多种参与铁稳态的基因或蛋白(包括铁的输入、输出及贮存)可以调节铁死亡的敏感性。固氮蛋白NFS1是一种半胱氨酸脱硫酶,它可以从半胱氨酸里为铁硫簇合成提供硫,激活铁饥饿应答,同时上调转铁蛋白受体的表达、下调铁蛋白FTH的表达,使细胞对铁死亡更敏感。溶酶体通过降解铁蛋白FTH,即所谓铁自噬,可以富集大量铁。抑制溶酶体功能或沉默核受体共激活因子NCOA4(一种负责将铁蛋白FTH招募到自噬小体从而使溶酶体降解铁蛋白、释放铁的运载蛋白)可以抑制铁死亡。过度激活HMOX1,催化亚铁血红素降解成二价铁离子、胆绿素和CO,可以通过增加不稳定铁池LIP进而促进铁死亡。利用withaferin A、erastin和BAY11-7085抑制或沉默HMOX1可以使铁死亡难以实施。不过,根据激活程度的不同,HMOX1也能在保护细胞方面发挥作用。HMOX1这种保护作用来自其抗氧化功能,而其细胞毒性是万一ferritin的缓冲作用不足时,它增强了亚铁离子的产生、促进了Fenton反应介导的过氧化物的分解。因此,过度上调HMOX1时有毒的,而适度上调HMOX1可能有保护细胞的作用。
脂质代谢
将原位合成的脂肪酸吸附进磷脂需要将长链脂肪酸转化成乙酰辅酶A,这一反应在乙酰辅酶A和溶血磷脂乙酰转移酶LPLATs的作用下完成。就这一点而言,敲低乙酰辅酶A合成酶长链家族成员4(ACSL4,可以将氨基酸转变成乙酰化的氨基酸),或丢失溶血卵磷脂乙酰转移酶3(LPCAT3,可催化乙酰化的氨基酸插入进磷脂中),都可以使细胞难发生铁死亡。最近,一种蛋白激酶级联的磷脂酰乙醇胺结合蛋白1(PEBP1)的支架蛋白抑制剂被发现可以结合、指导LOX15靶向细胞膜上的多不饱和脂肪酸PUFAs进而促进铁死亡。在研究FIN56诱导的铁死亡的激活机制时,甲羟戊酸盐通路被发现。甲羟戊酸盐的一个直接代谢产物异戊烯焦磷酸IPP对于胆固醇的生物合成、Sec-tRNA的异戊烯化以及CoQ10的产生。抑制鲨烯合成酶SQS和鲨烯单氧酶(参与胆固醇的合成,位于异戊烯焦磷酸的下游)的活性,可以阻碍铁死亡。
相反,用他汀类药物抑制HMG-CoA还原酶(位于异戊烯焦磷酸合成的上游)促进铁死亡。目前推测甲羟戊酸盐通路能通过2种机制影响铁死亡。Sec-tRNA是将丝氨酸Sec吸附进硒蛋白GPX4所必需的,抑制异戊烯焦磷酸合成会干扰Sec-tRNA的成熟。此外,抑制辅酶Q10的产生,可以造成线粒体不正常的呼吸功能和氧化损伤。就这一点而言,给细胞提供CoQ10可以有效抑制铁死亡。
抗氧化代谢
谷胱甘肽的代谢及其抗氧化能力调节对铁死亡的敏感性。通过分析60种人不同细胞系对铁死亡激活剂的敏感性,转录组数据发现NADPH丰富度作为一种生物标志,与对铁死亡诱导剂的敏感性负相关。转硫作用通路指的是,蛋氨酸通过从胱硫醚提供半胱氨酸用于谷胱甘肽的合成,当系统中Xc-被抑制时在一些细胞中调节铁死亡。上调对一些tRNA合成酶(CARS、HARS和EPRS)沉默能应答的转硫作用通路,可以使细胞对erastin诱导的铁死亡不敏感。抑制tRNA合成酶被认为可以诱导一种整合的压力应答,导致半胱氨酸合成的增强并最终促进GSH的合成。
值得注意的是,从药学方面抑制核受体NRF2、沉默NRF2相关基因可以增强细胞对铁死亡的敏感性,这反映出NRF2在诱导抗氧化机制方面的关键作用。用erastin和sorafenib刺激细胞可以提高NRF2的蛋白水平,使得细胞不易发生铁死亡。这种保护作用是通过上调NRF2的靶基因NQO1、HMOX1和FTH1实现的。线粒体是哺乳动物中氧自由基生成的来源。线粒体通过电子转运链产生氧自由基,进而导致铁死亡,但线粒体对于铁死亡并非是严格必需的。耗竭线粒体的细胞也能起始铁死亡。然而,抑制电子转运链复合体的活性与耗竭线粒体相比,是一种类似但是更近一步的途径,可以阻止半胱氨酸耗竭或erastin诱导的铁死亡,但是对GPX4抑制导致的铁死亡无效。
能量代谢
谷氨酸代谢,也就是谷氨酰胺分解,是半胱氨酸耗竭引起的铁死亡所必需的。抑制SLC1A5转运体对谷氨酰胺的摄取、抑制谷氨酰胺通过线粒体谷氨酰胺酶GLS2代谢成谷氨酸,以及阻止谷氨酸通过GOT1生成α-酮戊二酸,都会拮抗铁死亡。谷氨酰胺分解可能通过给线粒体三羧酸循环提供α-酮戊二酸调节了铁死亡。抑制戊糖磷酸化通路PPP或者沉默两种参与PPP的酶G6PD和PGD,阻碍了erastin在人肺癌细胞系中诱导的铁死亡。PPP通路产生NAPDH对于恢复细胞中的GSH水平、拮抗铁死亡是至关重要的。然而,在一些细胞中,通过提供NAPDH给NOXs,可以促进氧自由基的产生,促进铁死亡。
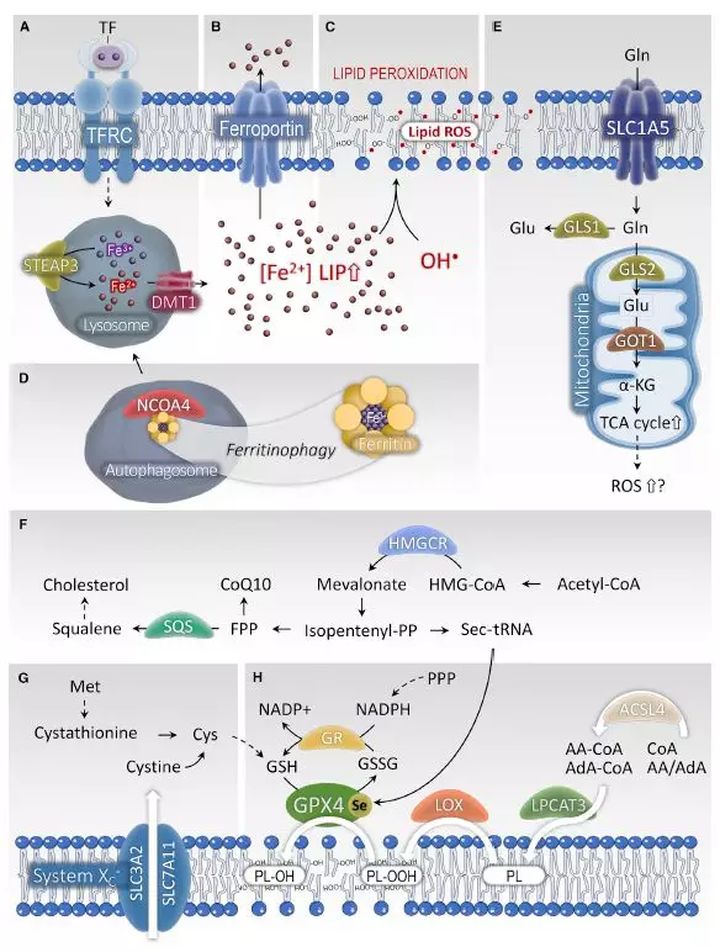
铁死亡和肿瘤
肿瘤的抑制分子,如BAP1和p53可以控制铁死亡的活性。此外,用一些小分子和FDA批准的药物在肿瘤细胞中激活铁死亡,在多种实验肿瘤模型中用铁死亡诱导剂抑制肿瘤的效果强调了铁死亡作为一种新的抗肿瘤治疗方案的潜力。而观察到的铁死亡可以杀死对治疗拮抗的肿瘤细胞进一步强化了这一方案的潜力。
BAP-1介导的铁死亡的调节
肿瘤抑制分子BAP1编码一种核去泛素化酶,存在于一种多梳-抑制去泛素酶复合体的形式,通过在核小体中减少组蛋白2A泛素化,从表观上调节基因的表达。BAP1突变见于多种散发的人类肿瘤中,其胚系突变被认为是遗传性肿瘤的重要诱因。BAP1的肿瘤抑制活性部分是通过SLC7A11启动子处H2A的去泛素化,使SLC7A11表达受到抑制,发生了铁死亡。体内实验发现,在BAP1缺陷的细胞中恢复BAP1的表达可以抑制异种移植的肿瘤的生长。人肿瘤相关BAP1突变不能抑制SLC7A11的表达,不能通过促进铁死亡抑制肿瘤。
P53介导的铁死亡的调节
TP53基因编码的基因组守护者p53是一个关键的肿瘤抑制子,在几乎一半的人类肿瘤中都是突变或失活的。肿瘤抑制子p53的活性主要与其经典的功能有关,包括细胞周期的滞留、细胞衰老或凋亡。然而,p53的非经典功能,比如控制代谢和氧化还原状态,也可以通过调节铁死亡抑制肿瘤。根据p53的突变状态和所处的细胞,针对氧化压力,p53具有促进铁死亡或拮抗铁死亡的功能。蛋白分子p53就像变阻器一样,在基础的或低水平的氧自由基压力下,具有抑制铁死亡的作用,而在高氧化压力状态下则促进铁死亡。
当处于细胞压力状态下,p53可以抑制SLC7A11转录、进而抑制胱氨酸摄取,使细胞易于发生铁死亡。例如,在骨肉瘤U2OS细胞中,存在氧自由基应激时,用nutlin-3激活p53可以起始铁死亡。调节SLC7A11的表达需要p53 DNA结合结构域的乙酰化。但是值得注意的是,突变p53上的3个赖氨酸位点(K177/161/162R,p533KR)使p53不能被乙酰化,可以使细胞不能发生凋亡、细胞衰老和细胞周期滞留,但细胞却更易发生铁死亡。这种p533KR敲入的小鼠不会形成自发肿瘤,这说明p53通过诱导铁死亡抑制肿瘤的形成。不同于p533KR,在p53敲除的小鼠中异位表达四乙酰化缺失突变的p534KR,不能在异位移植的模型中抑制SLC7A11,也不能抑制肿瘤的形成。P53也可以通过其代谢目标基因SAT1(其编码的蛋白参与的聚胺类代谢通路在人类肿瘤中通常是被下调的)调节铁死亡。敲除SAT1显著降低了p53介导的铁死亡的发生几率,而提高SAT1的表达水平使细胞面对氧自由基应激时更容易发生铁死亡。非洲人p53基因第47位丝氨酸(TP53S47)的遗传多态性特点会损害p53抑制SLC7A11转录的能力,使细胞不易发生铁死亡。虽然p53S47可以诱导凋亡、细胞衰老和细胞周期滞留,p53S47敲入的小鼠仍然易于发生自发性肿瘤。P53其它的突变形式(如p53R273H和p53R175H,来自TP53的错义突变)可以阻止NRF2介导的SLC7A11上调、抑制SLC7A11的表达。有趣的是,用p53G266R的激活剂APR-246激活p53G266R可以恢复柳氮磺胺吡啶对系统性Xc-的抑制,这在一个PDX模型(病人来源肿瘤异种动物模型)中抑制了肿瘤的生长。
在另一些细胞中,p53显然也可以负向调节铁死亡的发生。用nutlin-3稳定和恢复野生型p53的活性可以保护纤维肉瘤、肾癌和骨肉瘤细胞免于系统性Xc-的抑制(通过一种p53-21依赖轴维持细胞内谷胱甘肽的含量)诱发的铁死亡。这可以使肿瘤细胞在遭受代谢应激(如胱氨酸损失)时存活下来。另一中p53负向调节铁死亡的机制是p53可以阻断DPP4的活性,阻断erastin在结直肠癌细胞中诱导的铁死亡。当缺失p53时,DPP4与NOX1相互作用形成NOX1-DPP4复合体介导质膜脂质过氧化反应和铁死亡。
对治疗抵抗的肿瘤细胞对铁死亡的敏感性
对铁死诱导治疗抵抗的肿瘤细胞发生铁死亡是控制这些肿瘤细胞的一种好方法。例如,治疗抵抗的间叶细胞(以高表达ZEB1为特点)对于GPX4抑制剂和statin处理诱发的铁死亡高度敏感。ZEB1被认为是一种脂质生成因子并且可以调节脂质代谢,因此将间叶细胞基因表达和脂质过氧化的脆弱性联系了起来。类似地,耐药的存留细胞(persister cell)是肿瘤治疗的主要挑战,而它们的存活依赖GPX4,因此GPX4可以作为成为理想的靶标进而诱导铁死亡。在黑色素瘤异位移植模型中,GPX4还被证实是是肿瘤复发必要的因素。BRAF激酶抑制剂vemurafenib也可以使治疗抵抗的黑色素瘤细胞对许多激活剂诱导的铁死亡敏感。在治疗抵抗的好风险成神经细胞瘤中,铁死亡似乎也是有效的。用醉茄素A同时诱导经典和非经典的铁死亡可以有效杀死异质的高风险成神经细胞瘤细胞、抑制肿瘤生长;相较于依托泊苷和顺铂,在神经母细胞瘤异种移植的复发率方面表现更出色。
铁死亡诱导物治疗肿瘤的潜力
目前已经发现了四种起始铁死亡的方式。第一类铁死亡诱导物通过消耗谷胱甘肽诱导铁死亡,第二类铁死亡诱导物直接靶向灭活GPX4诱导铁死亡,第三类铁死亡诱导物通过SQS-me-valonate通路消耗GPX4和CoQ10,第四类铁死亡诱导物通过增加LIP或氧化铁诱导脂质过氧化反应。铁氧化物纳米颗粒可以通过提高铁和氧自由基的水平杀死肿瘤细胞。这使得纳米医学成为诱导铁死亡抗癌的新策略。
展望
自从发现了铁死亡,已经有强有力的证据显示:通过铁死亡可以在实验肿瘤模型中发挥抗肿瘤效果。治疗抵抗的肿瘤对铁死亡的脆弱性以及发现FDA批准的药物altretamine、sorafenib和硅纳米颗粒在肿瘤中可以作为铁死亡诱导剂,使我们对铁死亡治疗肿瘤的潜力产生了很高的期待。然而,还有很多问题需要进一步阐明。能用铁死亡进行治疗的肿瘤的特征是什么?在临床前和肿瘤临床治疗中,如何有效防控诱导铁死亡潜在的副作用?多大程度的肿瘤突变谱会影响肿瘤对铁死亡的敏感性?通过表观编辑,肿瘤会变得对铁死亡敏感吗?铁死亡的免疫原性怎样?诱导铁死亡和免疫疗法相比哪一个效果好?为了控制对铁死亡的敏感性,肿瘤细胞脂质代谢的可塑性是什么?
考虑到GPX4抑制剂在治疗抵抗肿瘤中的效果,开发和优化靶向GPX4的化合物将面临改善药代动力学和靶向性的挑战。从这个方面来说,纳米医学因其更好的效果和提高的靶向性降低了系统毒性、提高了安全性,成为开发铁死亡的好方案。除了抗肿瘤的作用,铁死亡的生理功能是什么?铁死亡为什么是进化保守的?脂质过氧化反应在多大程度上与铁死亡功能相关?脂质过氧化反应是膜透化的原因吗?脂质加合物诱导的膜蛋白修饰对于通道、孔或受体的功能改变是必需的吗?现在大多数机制研究中用到的脂质过氧化物荧光检测方法是否能精确测定细胞脂质的过氧化物?亲脂自由基诱饵对于控制脂质氧自由基、脂质过氧化物和铁死亡诱导的损伤有什么治疗潜力?为了提高对难治肿瘤的应答,更详尽的机制研究和实时铁死亡诊断工具是必需的。
原文链接: