GTEx(Genotype-Tissue Expression,基因型-组织表达)数据库,研究从来自449名生前健康的人类捐献者的7000多份尸检样本,涵盖44个组织(42种不同的组织类型),包括31个实体器官组织、10个闹分区、全血、2个来自捐献者血液和皮肤的细胞系,作者利用这些样本研究基因表达在不同组织和个体中有何差异。
GTEx对几乎所有转录基因的基因表达模式进行了观察,从而能够确定基因组中影响基因表达的特定区域。
此外,合并GTEx与TCGA数据库数据能够有效解决TCGA数据库中正常组织样本量不足的缺陷,从而提高比较的准确性。
前面代码是从完整表达数据开始整理,数据太大,没有必要,大家可以学习一下,针对单基因,代码在后面B站视频那一块,很方便!
1. 数据来源
- tcga_RSEM_gene_tpm.gz
- TCGA_phenotype_denseDataOnlyDownload.tsv.gz
- gtex_RSEM_gene_tpm.gz
- GTEX_phenotpye.gz
- gencode.v23.annotation.gene.probemap
- samplepair.txt(TCGA和GTeX sample信息)
(数据比较大,如果下载困难可以留言)
2. 注释来自 TCGA 和 GTEx 的样本
library(stringr)
library(dplyr)
library(ggplot2)
library(RColorBrewer)
library(data.table)
#################======= step1: clean GTEx pheno data =======#################
gtex <- read.table("samplepair.txt",header=T,sep='\t')
tcga_ref <- gtex[,1:2]
gtex$type <- paste0(gtex$TCGA,"_normal_GTEx")
gtex$sample_type <-"normal"
gtex <- gtex[,c("TCGA","GTEx","type","sample_type")]
names(gtex)[1:2] <- c("tissue","X_primary_site")
gp <- read.delim(file="GTEX_phenotype.gz",header=T,as.is = T)
gtex2tcga <- merge(gtex,gp,by="X_primary_site")
gtex_data <- gtex2tcga[,c(5,2:4)]
names(gtex_data)[1] <- "sample"
#write.table(gtex_data,"GTEx_pheno.txt",row.names=F,quote=F,sep='\t')
#################======= step2: clean a TCGA pheno data =======#################
tcga <- read.delim(file="TCGA_phenotype_denseDataOnlyDownload.tsv.gz",header=T,as.is = T)
tcga <- merge(tcga_ref,tcga,by.y="X_primary_disease",by.x="Detail",all.y = T)
tcga <- tcga[tcga$sample_type %in% c("Primary Tumor","Solid Tissue Normal"),]
tcga$type <- ifelse(tcga$sample_type=='Solid Tissue Normal',
paste(tcga$TCGA,"normal_TCGA",sep="_"),paste(tcga$TCGA,"tumor_TCGA",sep="_"))
tcga$sample_type <- ifelse(tcga$sample_type=='Solid Tissue Normal',"normal","tumor")
tcga<-tcga[,c(3,2,6,5)]
names(tcga)[2] <- "tissue"
#write.table(tcga,"tcga_pheno.txt",row.names = F,quote=F,sep='\t')
#################======= step3: remove samples without tpm data =======############
gtex_exp <- fread("gtex_RSEM_gene_tpm.gz",data.table = F)
gtexS <- gtex_data[ gtex_data$sample%in%colnames(gtex_exp)[-1],]
tcga_exp <- fread("tcga_RSEM_gene_tpm.gz",data.table = F)
tcgaS <- tcga[tcga$sample %in%colnames(tcga_exp)[-1],]
tcga_gtex <- rbind(tcgaS,gtexS)
write.table(tcga_gtex,"tcga_gtex_sample.txt",row.names = F,quote=F,sep='\t')
3. 提取感兴趣的基因
library(stringr)
library(dplyr)
library(ggplot2)
library(RColorBrewer)
library(data.table)
library(tibble)
rm(list = ls())
options(stringsAsFactors = FALSE)
target <- "YTHDC2"
idmap <- read.delim("gencode.v23.annotation.gene.probemap",as.is=T)
tcga_exp <- fread("tcga_RSEM_gene_tpm.gz",data.table = F)
gtex_exp <- fread("gtex_RSEM_gene_tpm.gz",data.table=F)
tcga_gtex <- read.table("tcga_gtex_sample.txt",sep='\t',header = T)
id <- idmap$id[which(idmap$gene==target)]
tcga_data <- t(tcga_exp[tcga_exp$sample==id,colnames(tcga_exp)%in%c("sample",tcga_gtex$sample)])
tcga_data <- data.frame(tcga_data[-1,])
tcga_data <- rownames_to_column(tcga_data,"sample")
names(tcga_data)[2] <- "tpm"
gtex_data <- t(gtex_exp[gtex_exp$sample==id,colnames(gtex_exp)%in%c("sample",tcga_gtex$sample)])
gtex_data <- data.frame(gtex_data[-1,])
gtex_data <- rownames_to_column(gtex_data,"sample")
names(gtex_data)[2] <- "tpm"
tmp <- rbind(tcga_data,gtex_data)
exp <- merge(tmp,tcga_gtex,by="sample",all.x=T)
exp <- exp[,c("tissue","sample_type","tpm")]
exp <- arrange(exp,tissue)
write.table(exp,"Merge gene expression/YTHDC2 expression.txt",row.names = F,quote=F,sep='\t')
4. 可视化基因表达
library(ggplot2)
library(ggpubr)
library(RColorBrewer)
rm(list = ls())
options(stringsAsFactors = FALSE)
exp <- read.table("Merge gene expression/YTHDC2 expression.txt",header=T,sep='\t')
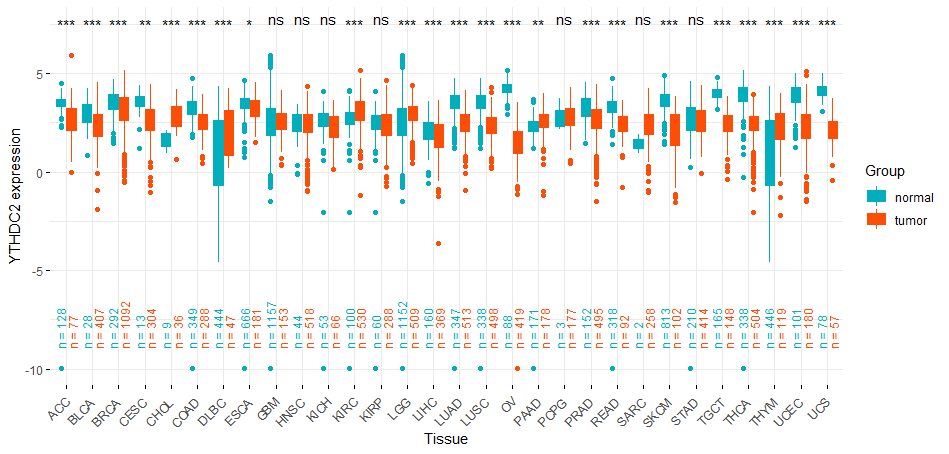
ylabname <- paste("YTHDC2", "expression")
colnames(exp) <- c("Tissue", "Group", "Gene")
p1 <- ggboxplot(exp, x = "Tissue", y = "Gene", fill = 'Group',
ylab = ylabname,
color = "Group",
palette = c("#00AFBB", "#FC4E07"),
ggtheme = theme_minimal())
##计算每种肿瘤正常和肿瘤组织的样本量
count_N<-exp %>% group_by(Tissue, Group) %>% tally
count_N$n <- paste("n =",count_N$n)
##添加N = 到图中
p1 <-p1+geom_text(data=count_N, aes(label=n, y=-9,color=Group), position=position_dodge2(0.9),size = 3,angle=90, hjust = 0)+
theme(axis.text.x = element_text(angle = 45,hjust = 1.2))
#计算t检验显著性
comp<- compare_means(Gene ~ Group, group.by = "Tissue", data = exp,
method = "t.test", symnum.args = list(cutpoints = c(0,0.001, 0.01, 0.05, 1), symbols = c( "***", "**", "*", "ns")),
p.adjust.method = "holm")
#添加显著性标记
p2 <- p1 + stat_pvalue_manual(comp, x = "Tissue", y.position = 7.5,
label = "p.signif", position = position_dodge(0.8))
p2
#dev.off()
##保存图片
### pdf version
ggsave("figure/pancancer_Plot.pdf", width = 14, height = 5)
### png version
#png("figure/pancancer_Plot.png", width = 465, height = 225, units='mm', res = 300)
代码参考GitHub:https://github.com/cmutd/TCGA_GTEx
B站视频更新代码(从Xena下载的数据),其余代码一样(tcga_gtex.rda样本注释文件关注公众号下载,公众号二维码在导航栏:科研互助–>B站代码获取,发送tcga_gtex即可,网盘链接电脑浏览器打开即可下载,不需要下载客户端):
load("tcga_gtex.rda")
exp <- read.table("YTHDC2.tsv",header=T,sep='\t')
exp <- exp[c(1,3)]
exp <- merge(exp, tcga_gtex,by="sample")
colnames(exp)[c(2,3,5)] <- c("Gene","Tissue","Group")
ylabname <- paste("YTHDC2", "expression")
exp <- exp %>% plotly::filter(Gene != -9.966)
library(ggpubr)
p1 <- ggboxplot(exp, x = "Tissue", y = "Gene", fill = 'Group',
ylab = ylabname,
color = "Group",
palette = c("#00AFBB", "#FC4E07"),
ggtheme = theme_minimal())
##计算每种肿瘤正常和肿瘤组织的样本量
count_N<-exp %>% group_by(Tissue, Group) %>% tally
count_N$n <- paste("n =",count_N$n)
##添加N = 到图中
p1 <-p1+geom_text(data=count_N, aes(label=n, y=-9,color=Group), position=position_dodge2(0.9),size = 3,angle=90, hjust = 0)+
theme(axis.text.x = element_text(angle = 45,hjust = 1.2))
#计算t检验显著性
comp<- compare_means(Gene ~ Group, group.by = "Tissue", data = exp,
method = "t.test", symnum.args = list(cutpoints = c(0,0.001, 0.01, 0.05, 1), symbols = c( "***", "**", "*", "ns")),
p.adjust.method = "holm")
#添加显著性标记
p2 <- p1 + stat_pvalue_manual(comp, x = "Tissue", y.position = 7.5,
label = "p.signif", position = position_dodge(0.8))
p2
tcga_gtex.rda文件获取—关注公众号之后,网盘链接没办法用浏览器打开,请问是不是更换了呀
内容: tcga_gtex.rda
点击链接查看: https://console.zbox.filez.com/l/W0TBNs
提取码: ejbj
到期日: 2030-03-28
你好,“tcga_gtex.rda样本注释文件”在公众号的链接显示已失效,请问怎么下载呢?
对于合并后的数据去批次之后出现负数还能用limma分析吗
进哥,您公众号的tcga_gtex.rda样本注释文件失效了,可以麻烦您再分享一份吗
博主您好,我从GTEx下载了表达矩阵后,进行2^exp-0.001还原,还原后的基因表达仍然有小于零的负值是为什么?这种情况应该如何处理呢?
大佬,你好,请问这个数据【TCGA_phenotype_denseDataOnlyDownload.tsv.gz 】从哪里生成或下载的呢?UCSC还是GTEX呢,想了解一下简单的原理
进哥你好,如果我按照你一开始下载原始表达数据的代码进行TCGA_GTEx联合分析,而不是从Xnea直接下载三个数据库的单基因表达数据(因为我不想加上TARGET数据库)。我看你代码里没有进行去批次化,直接合并的,请问这个需要去批次化嘛?感谢进哥!
##计算每种肿瘤正常和肿瘤组织的样本量
count_N% group_by(Tissue, Group) %>% tally
count_N$n <- paste("n =",count_N$n)
进哥我的这个代码提示我
错误于tally(.): 没有"tally"这个函数
怎么解决
进哥您好,我想问下前面2步下载tcga和gtex的那4个数据,为什么还用后面的那个ucsc网站再下数据合并呢?是使用了那个网站下好了基因就不需要前面下载那四个数据并进行数据整理了么?
你好进哥,运行到最后显示这个怎么解决
Error in `mutate()`:
ℹ In argument: `p = purrr::map(…)`.
Caused by error in `purrr::map()`:
ℹ In index: 26.
ℹ With name: Tissue.CESC.
Caused by error in `t.test.default()`:
! ‘y’观察值数量不够
—
王老师,我想问一下TARGET和TCGA数据合并分析肿瘤组织中的表达可以做嘛?
老师:你好,GTEX数据是RSEM count,而TCGA是STAR count,可以直接合并后做差异分析吗?谢谢!
王博,你好,看到你这篇文章对我的帮助很大,但是我在做肝癌时用的UCSX的TCGA target GTEX数据时,根据肿瘤与非肿瘤分组绘制PCA时,发现还是存在着批次效应,感觉很奇怪
王博,你好,看到你这篇文章对我的帮助很大,但是我在做肝癌时用的UCSX的TCGA target GTEX数据时,根据肿瘤与非肿瘤分组绘制PCA时,发现还是存在着批次效应,感觉很奇怪
你好 我想问一下 按照上述代码运行后 P1跑出来没有图片是什么原因呢,有什么办法可以解决吗?打扰了
大大,数据太大很难下载下来怎么办?gtex_RSEM_gene_tpm.gz 这个下载速度只有几kb还一直下载失败?请问该怎么解决?
博主你好,可以分享一下samplepair.txt这个文件吗?
王老师你好,我在运行了p1 <- ggboxplot(exp, x = "Tissue", y = "Gene", fill = 'Group',
ylab = ylabname,
color = "Group",
palette = c("#00AFBB", "#FC4E07"),
ggtheme = theme_minimal())这段代码后plot窗口没能显示图片,再试验了其他简单的作图代码后可以正常显示,而且前面几部运行结果也是正常的,输出结果和您视频里演示的结果相似,请问该怎么解决呀
Error in `train_discrete()`:
! Continuous value supplied to a discrete scale
这个问题怎么解决啊,在运行可视化基因表达的时候出现的
王老师,您好。我想请教下ICGC数据库中下载数据能直接和GTEX数据库数据联合使用么?
王老师,您好。我想请教下 tcga_RSEM_gene_tpm.gz 这个数据从哪儿下载的,如果我想换成count数据怎么办?
> #计算t检验显著性
> comp<- compare_means(Gene ~ Group, group.by = "Tissue", data = exp,
+ method = "t.test", symnum.args = list(cutpoints = c(0,0.001, 0.01, 0.05, 1), symbols = c( "***", "**", "*", "ns")),
+ p.adjust.method = "holm")
Error in (function (x, cutpoints = c(0.3, 0.6, 0.8, 0.9, 0.95), symbols = if (numeric.x) c(" ", :
argument "x" is missing, with no default
In addition: Warning message:
Unknown or uninitialised column: `p`.
你好,在跑的过程中出现以上错误该怎么解决呀
进哥,GTEx中的数据处理显示是log2(tpm+0.01),而TCGA中的数据处理是log2(tpm+1),这在合并时需要将GTEx中的数据还原再进行log2(tpm+1)的处理 吗?
进哥哥,GTEx数据库进不了啊?用了vpn也没有用。
进哥,我想问一下plotly这个包怎么下载啊?exp % plotly::filter(Gene != -9.966)运行这句代码的时候报错没有plotly这个包
哥哥,ggboxplot中怎么显示箱子的轮廓,上四分位数,中位数,下四分位数呢?
up您好,请问您能单独出一期数据的下载吗,我把您b站上的视频都看了,tcga_gtex、samplepair等数据的下载没有教程,想学习一下
检查了一下,第一发现下载UCSC中TCGA和GTEX数据中某个基因TPM表达量时,虽然选择的时候至少选择两种以上,但下载表达矩阵的时候可以只下载TPM。第二个问题是在运行R代码时,tsv文件的第三列是FPKMZ值,而第四列为TPM值,但代码中提取的是第一列和第三列。视频里说的是提取TPM,实际提取的为FPKM。这点需要更正一下
进哥您好,我想问一下,为什么TCGA_phenotype_denseDataOnlyDownload.tsv.gz点开之后是乱码,下载不了
进哥您好,请问出现Error in file(file, ifelse(append, “a”, “w”)) :
cannot open the connection
In addition: Warning message:
In file(file, ifelse(append, “a”, “w”)) :
cannot open file ‘Merge gene expression/MCMBP expression.txt’: No such file or directory是什么原因呢,谢谢您
王博您好,你的这个教程十分受用,非常棒。我用同样的代码换个基因运行后出现一个问题,部分肿瘤正常组织箱式图的75%下限位于-10以下,图片中不显示出来。这个是什么原因呢?
您好,基因的表达太低,又有些缺失值,在这个数据里面也就是-9.9658,删掉看看
进哥能详细讲述一下文章开头处数据来源如何下载的吗?
XENA上下载的,也有链接,我B站上有更加方便的方法,就是最下面的视频,你看看先
您好,这个文件( samplepair.txt)在那儿里下载呀,我下载不下载fpkm值改成count值可以吗
谢谢!
您好,请问这里是否需要去除批次效应?
3大数据库超2万RNA-seq数据重新统一处理——关于TCGA-GTEx是否需要标准化 – 王进的个人网站
https://www.jingege.wang/2023/05/24/3%e5%a4%a7%e6%95%b0%e6%8d%ae%e5%ba%93%e8%b6%852%e4%b8%87rna-seq%e6%95%b0%e6%8d%ae%e9%87%8d%e6%96%b0%e7%bb%9f%e4%b8%80%e5%a4%84%e7%90%86-%e5%85%b3%e4%ba%8etcga-gtex%e6%98%af%e5%90%a6/
这里有解释过的,已经是处理过后的数据
博主,您好,我想问一下TCGA 和 GTEx 样本的TPM在做差异分析之前是否需要进行去批次效应,我这边进行去批次效应后表达出现了负值,无法进行后续分析
3大数据库超2万RNA-seq数据重新统一处理——关于TCGA-GTEx是否需要标准化 – 王进的个人网站
https://www.jingege.wang/2023/05/24/3%e5%a4%a7%e6%95%b0%e6%8d%ae%e5%ba%93%e8%b6%852%e4%b8%87rna-seq%e6%95%b0%e6%8d%ae%e9%87%8d%e6%96%b0%e7%bb%9f%e4%b8%80%e5%a4%84%e7%90%86-%e5%85%b3%e4%ba%8etcga-gtex%e6%98%af%e5%90%a6/
已经是去过批次的,另外有负值也是正常的 用的是log转化的数据
进哥您好,萌新一枚,请问我运行您视频第54行代码时提示Error in ggboxplot(exp, x = “Tissue”, y = “Gene”, fill = “Group”, ylab = ylabname, :
could not find function “ggboxplot”
请问是什么原因呢
解决啦,谢谢进哥
加载一下那个包library(ggpubr)
进哥您好,我运行您视频中得第54行时提示Error in ggboxplot(exp, x = “Tissue”, y = “Gene”, fill = “Group”, ylab = ylabname, : could not find function “ggboxplot”请问是什么原因呢
library(ggpubr)
加载一下包
您好,请问差异分析不是一般用counts数据么?为什么这里用的是tpm数据呢?
不是呀,Count是没有进行标准化的数据,对于Count,用DESeq2进行差异分析
如果获得的是TPM或FPKM,也可以用Limma进行差异分析
对于单个基因的分析,秩和检验或t检验都可以,这个针对你的分析要求决定,方法中说明即可
您好,用wilcox分析差异OK吗?GEPIA2使用的ANOVA分析和wilcox差异很大,不确定哪个方法结果可信。
大样本量需不需要f检验啊
博主您好,想问一下评论里说的按肿瘤分割好的网盘链接在哪里呀?我似乎没有找到网盘的链接。
底下old comments
重新发一下:https://pan.baidu.com/s/17blyTZb-Kni8u9yqIsweyA?pwd=eqsp
提取码:eqsp
非常感谢
王博士您好,我想问一下如果那个样本信息是如何得到的呢?
您好,请问指的是这个吗?https://xenabrowser.net/datapages/?dataset=TCGA_GTEX_category.txt&host=https%3A%2F%2Ftoil.xenahubs.net&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443
您看一下
感谢您的回复~我想应该不是这个,是这个文件里面的信息samplepair.txt。
OK 这个可以自己下载每个癌症的样本信息 自己整合
请问网盘链接是被删除了么,没有看到
前面文件名上直接到xena的下载链接
请问一下,看您的数据用的tpm格式的,差异分析的时候不是一般用count值吗,为什么这里用了tpm呢。谢谢您的解答
请问,这些数据是从USCS上下载的吗,但是我看着说这上面数据更新比较慢,和tcga数据库比数据可能不一样
是的 存在滞后,如果一定需要最新数据进行分析,那就需要自行下载了
要如何把单一肿瘤的数据拆分出来
底下评论有网盘链接 我分割好的
博主,您好,我自己写代码分析了Gtex和TCGA数据库的数据。首先将二者转换成TPM数据类型,然后对二者分别进行了normalize,接着进行了去批次,最后是对数据进行了调整,绘制单基因泛癌表达。但得到的部分样本的表达量小于0,是负值。然后网上有幸搜到您的代码,自己运行了一遍,发现跑出来的结果和我的类似,也会有一部分样本的表达量为负值,这样的图觉得很奇怪。请问,这种情况应该怎么调整?
您好,如果使用的是log2转换之后的数据,必然会有负值,也就是TPM介于0-1的,这是正常的,而TPM本身不会有负值
实际TPM=0的经过log2(TPM+0.001)之后数值为-9.8.。。。如果不想纳入表达为0的 可以将这些数据删掉
那请问下 可不可以直接设定y的坐标范围大于零呢?