GTEx(Genotype-Tissue Expression,基因型-组织表达)数据库,研究从来自449名生前健康的人类捐献者的7000多份尸检样本,涵盖44个组织(42种不同的组织类型),包括31个实体器官组织、10个闹分区、全血、2个来自捐献者血液和皮肤的细胞系,作者利用这些样本研究基因表达在不同组织和个体中有何差异。
GTEx对几乎所有转录基因的基因表达模式进行了观察,从而能够确定基因组中影响基因表达的特定区域。
此外,合并GTEx与TCGA数据库数据能够有效解决TCGA数据库中正常组织样本量不足的缺陷,从而提高比较的准确性。
前面代码是从完整表达数据开始整理,数据太大,没有必要,大家可以学习一下,针对单基因,代码在后面B站视频那一块,很方便!
1. 数据来源
- tcga_RSEM_gene_tpm.gz
- TCGA_phenotype_denseDataOnlyDownload.tsv.gz
- gtex_RSEM_gene_tpm.gz
- GTEX_phenotpye.gz
- gencode.v23.annotation.gene.probemap
- samplepair.txt(TCGA和GTeX sample信息)
(数据比较大,如果下载困难可以留言)
2. 注释来自 TCGA 和 GTEx 的样本
library(stringr)
library(dplyr)
library(ggplot2)
library(RColorBrewer)
library(data.table)
#################======= step1: clean GTEx pheno data =======#################
gtex <- read.table("samplepair.txt",header=T,sep='\t')
tcga_ref <- gtex[,1:2]
gtex$type <- paste0(gtex$TCGA,"_normal_GTEx")
gtex$sample_type <-"normal"
gtex <- gtex[,c("TCGA","GTEx","type","sample_type")]
names(gtex)[1:2] <- c("tissue","X_primary_site")
gp <- read.delim(file="GTEX_phenotype.gz",header=T,as.is = T)
gtex2tcga <- merge(gtex,gp,by="X_primary_site")
gtex_data <- gtex2tcga[,c(5,2:4)]
names(gtex_data)[1] <- "sample"
#write.table(gtex_data,"GTEx_pheno.txt",row.names=F,quote=F,sep='\t')
#################======= step2: clean a TCGA pheno data =======#################
tcga <- read.delim(file="TCGA_phenotype_denseDataOnlyDownload.tsv.gz",header=T,as.is = T)
tcga <- merge(tcga_ref,tcga,by.y="X_primary_disease",by.x="Detail",all.y = T)
tcga <- tcga[tcga$sample_type %in% c("Primary Tumor","Solid Tissue Normal"),]
tcga$type <- ifelse(tcga$sample_type=='Solid Tissue Normal',
paste(tcga$TCGA,"normal_TCGA",sep="_"),paste(tcga$TCGA,"tumor_TCGA",sep="_"))
tcga$sample_type <- ifelse(tcga$sample_type=='Solid Tissue Normal',"normal","tumor")
tcga<-tcga[,c(3,2,6,5)]
names(tcga)[2] <- "tissue"
#write.table(tcga,"tcga_pheno.txt",row.names = F,quote=F,sep='\t')
#################======= step3: remove samples without tpm data =======############
gtex_exp <- fread("gtex_RSEM_gene_tpm.gz",data.table = F)
gtexS <- gtex_data[ gtex_data$sample%in%colnames(gtex_exp)[-1],]
tcga_exp <- fread("tcga_RSEM_gene_tpm.gz",data.table = F)
tcgaS <- tcga[tcga$sample %in%colnames(tcga_exp)[-1],]
tcga_gtex <- rbind(tcgaS,gtexS)
write.table(tcga_gtex,"tcga_gtex_sample.txt",row.names = F,quote=F,sep='\t')
3. 提取感兴趣的基因
library(stringr)
library(dplyr)
library(ggplot2)
library(RColorBrewer)
library(data.table)
library(tibble)
rm(list = ls())
options(stringsAsFactors = FALSE)
target <- "YTHDC2"
idmap <- read.delim("gencode.v23.annotation.gene.probemap",as.is=T)
tcga_exp <- fread("tcga_RSEM_gene_tpm.gz",data.table = F)
gtex_exp <- fread("gtex_RSEM_gene_tpm.gz",data.table=F)
tcga_gtex <- read.table("tcga_gtex_sample.txt",sep='\t',header = T)
id <- idmap$id[which(idmap$gene==target)]
tcga_data <- t(tcga_exp[tcga_exp$sample==id,colnames(tcga_exp)%in%c("sample",tcga_gtex$sample)])
tcga_data <- data.frame(tcga_data[-1,])
tcga_data <- rownames_to_column(tcga_data,"sample")
names(tcga_data)[2] <- "tpm"
gtex_data <- t(gtex_exp[gtex_exp$sample==id,colnames(gtex_exp)%in%c("sample",tcga_gtex$sample)])
gtex_data <- data.frame(gtex_data[-1,])
gtex_data <- rownames_to_column(gtex_data,"sample")
names(gtex_data)[2] <- "tpm"
tmp <- rbind(tcga_data,gtex_data)
exp <- merge(tmp,tcga_gtex,by="sample",all.x=T)
exp <- exp[,c("tissue","sample_type","tpm")]
exp <- arrange(exp,tissue)
write.table(exp,"Merge gene expression/YTHDC2 expression.txt",row.names = F,quote=F,sep='\t')
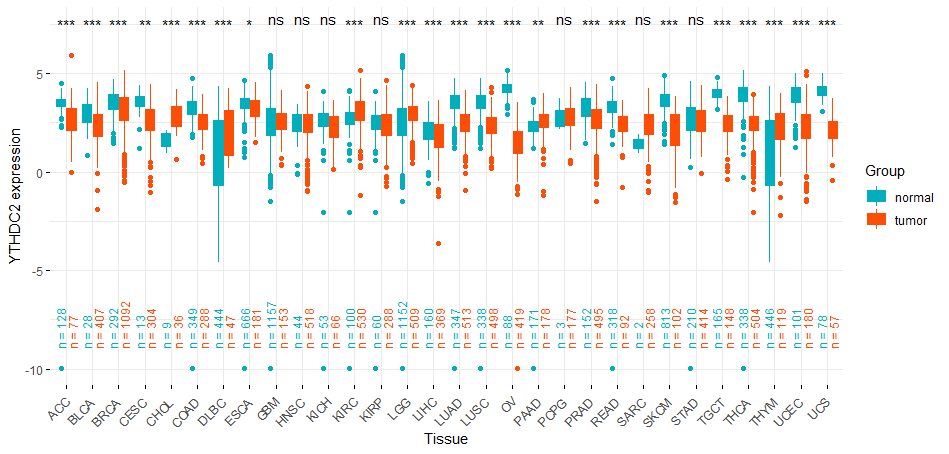
4. 可视化基因表达
library(ggplot2)
library(ggpubr)
library(RColorBrewer)
rm(list = ls())
options(stringsAsFactors = FALSE)
exp <- read.table("Merge gene expression/YTHDC2 expression.txt",header=T,sep='\t')
ylabname <- paste("YTHDC2", "expression")
colnames(exp) <- c("Tissue", "Group", "Gene")
p1 <- ggboxplot(exp, x = "Tissue", y = "Gene", fill = 'Group',
ylab = ylabname,
color = "Group",
palette = c("#00AFBB", "#FC4E07"),
ggtheme = theme_minimal())
##计算每种肿瘤正常和肿瘤组织的样本量
count_N<-exp %>% group_by(Tissue, Group) %>% tally
count_N$n <- paste("n =",count_N$n)
##添加N = 到图中
p1 <-p1+geom_text(data=count_N, aes(label=n, y=-9,color=Group), position=position_dodge2(0.9),size = 3,angle=90, hjust = 0)+
theme(axis.text.x = element_text(angle = 45,hjust = 1.2))
#计算t检验显著性
comp<- compare_means(Gene ~ Group, group.by = "Tissue", data = exp,
method = "t.test", symnum.args = list(cutpoints = c(0,0.001, 0.01, 0.05, 1), symbols = c( "***", "**", "*", "ns")),
p.adjust.method = "holm")
#添加显著性标记
p2 <- p1 + stat_pvalue_manual(comp, x = "Tissue", y.position = 7.5,
label = "p.signif", position = position_dodge(0.8))
p2
#dev.off()
##保存图片
### pdf version
ggsave("figure/pancancer_Plot.pdf", width = 14, height = 5)
### png version
#png("figure/pancancer_Plot.png", width = 465, height = 225, units='mm', res = 300)
代码参考GitHub:https://github.com/cmutd/TCGA_GTEx
B站视频更新代码(从Xena下载的数据),其余代码一样(tcga_gtex.rda样本注释文件关注公众号下载,公众号二维码在导航栏:科研互助–>B站代码获取,发送tcga_gtex即可,网盘链接电脑浏览器打开即可下载,不需要下载客户端):
load("tcga_gtex.rda")
exp <- read.table("YTHDC2.tsv",header=T,sep='\t')
exp <- exp[c(1,3)]
exp <- merge(exp, tcga_gtex,by="sample")
colnames(exp)[c(2,3,5)] <- c("Gene","Tissue","Group")
ylabname <- paste("YTHDC2", "expression")
exp <- exp %>% plotly::filter(Gene != -9.966)
library(ggpubr)
p1 <- ggboxplot(exp, x = "Tissue", y = "Gene", fill = 'Group',
ylab = ylabname,
color = "Group",
palette = c("#00AFBB", "#FC4E07"),
ggtheme = theme_minimal())
##计算每种肿瘤正常和肿瘤组织的样本量
count_N<-exp %>% group_by(Tissue, Group) %>% tally
count_N$n <- paste("n =",count_N$n)
##添加N = 到图中
p1 <-p1+geom_text(data=count_N, aes(label=n, y=-9,color=Group), position=position_dodge2(0.9),size = 3,angle=90, hjust = 0)+
theme(axis.text.x = element_text(angle = 45,hjust = 1.2))
#计算t检验显著性
comp<- compare_means(Gene ~ Group, group.by = "Tissue", data = exp,
method = "t.test", symnum.args = list(cutpoints = c(0,0.001, 0.01, 0.05, 1), symbols = c( "***", "**", "*", "ns")),
p.adjust.method = "holm")
#添加显著性标记
p2 <- p1 + stat_pvalue_manual(comp, x = "Tissue", y.position = 7.5,
label = "p.signif", position = position_dodge(0.8))
p2
进哥哥,求一份三阴乳腺癌的TCGA+GTEx整合的表达矩阵文件,及临床信息,还想要三阴乳腺癌的TCGA+GTEx数据
我没有提取三阴性的数据,整个的BLCA的有的,你可以从留言的网盘下载,然后依据临床数据提取三阴性的数据
不清楚的话可以加微信 告诉你怎么弄
进哥哥,你好!
我想联合TCGA与GTEx做结肠癌的差异分析,我看网络视频,使用的UCSC中的fpkm数据,两者合并后用limma包,做差异分析。
我想请教的问题是:用limma包做差异分析,可以使用FPKM数据马?我看网上好多人说必须用counts值,而我用的fpkm,心里好慌。
望,进哥哥,多多指导,谢谢!
limma可以分析log转化之后的TPM或FPKM吗,deseq2必须count,当然最好用count进行所有的差异分析
详细可以参考官方说明,测序数据经过标准化之后可以用limma进行进一步差异分析,可以接受
关于tpm的差异表达分析,limma包作者Gordon K. Smyth是这么说的:
In my opinion, there is no good way to do a DE analysis of RNA-seq data starting from the TPM values. TPMs just throw away too much information about the original count sizes. Sorry, but I’m not willing to make any recommendations, except to dissuade people from thinking that TPMs are an adequate summary of an RNA-seq experiment.
I see that some people in the literature have done limma analyses of the log(TPM+1) values and, horrible though that is, I can’t actually think of anything better, given TPMs and existing software. One could make this a little better by using eBayes with trend=TRUE and by using arrayWeights() to try to partially recover the library sizes. Please do not take that as a recommendation though!
进哥哥,下午好!
您这边有合适代码,基于结肠癌COAD,将GTEx联合TCGA数据的counts矩阵提取出来吗?我想做结肠癌的差异分析。
望进哥哥多多提点,谢谢!
您好,我有看到有人评论说FPKM可以用非参数做差异分析,请问这个可行性怎么样?
可以呀 但是一般不建议用fpkm 建议tpm更好
你好
进哥哥
我想请教一下,网上都说TPM是标准化的数据,可以用来进行差异分析,这是不是直接进行差异分析即可(比如用SPSS等分析软件进行分析),而limma是对count数据进行标准化后再做差异分析的,所以只能用count数据,我这样理解对吗?
另外我想请教下,代码中并没有去除批次效应,这样的数据可以进行差异分析吗
3大数据库超2万RNA-seq数据重新统一处理——关于TCGA-GTEx是否需要标准化
limma主要针对芯片数据,对于tpm,官网说明也是可以的;如果拿count分析,需要DEseq2先进行标准化,然后limma、DEseq2和EdgeR均可用于后续分析
您好,进哥哥,我在GTEx网站下载的counts不知道为什么都不是整数,可以用DEseq2标准化吗?步骤是怎样的呀?
您好,我想问下GTEx数据库中是没有直肠部位的数据吗?
您好 大概看了 好像只有colon 没有直肠
博主您好,向求一份GTEX联合TCGA肺腺癌合并后的表达矩阵,可以吗?跪谢
您好 底下链接有单独分割好的数据 我在这里贴上代码 你试试看,有问题再讨论
lung <- read.csv("G:/GTEx/Lung.csv") luad <- read.csv("G:/TCGA Pancancer/split/LUAD.csv") lung[1:4,1:4] luad[1:4,1:4] merge_data <- merge(lung,luad,by = "X") write.csv(merge_data,"LUAD_GTEx.csv",row.names = F) ID <- colnames(merge_data)[-1] group <- data.frame(ID = ID, Group = ifelse(substr(ID,13,15) == ".01", "Tumor", "Normal")) write.csv(group,"Group.csv",row.names = F)
进哥,求一份胰腺癌TCGA和GTEx的合并矩阵可以吗
自己合并好了吗 之前留言应该有代码 搞不定的话加我微信
跪求TCGA联合GTEX的肺腺癌的合并矩阵
https://pan.baidu.com/s/17blyTZb-Kni8u9yqIsweyA?pwd=eqsp
提取码:eqsp
下载后用我发你的代码合并 生成分组文件
您好,我在分析低级别胶质瘤数据时对GTEX和TCGA数据进行了合并,来对比癌症患者样本和正常组织样本中相关基因表达差异。然而收到的反馈意见是,编辑认为在GTEx中有多种脑组织数据,存在批次效应,目前尚不清楚作者是如何合并来自不同脑区的转录组数据。请问一下我这种情况有无解决途径。
这个真不好解决,不过你可以使用这个数据:https://xenabrowser.net/datapages/?dataset=TcgaTargetGtex_rsem_gene_tpm&host=https%3A%2F%2Ftoil.xenahubs.net&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443
是合并好的,来源如下:
Toil enables reproducible, open source, big biomedical data analyses – PMC
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5546205/
请问您批次效应的问题最后怎么解决的?
您好,请教个问题,请问下合并好的tpm数据有负值,令负值为缺失值NA,接下来用什么代码去删除含有NA的行呢?尝试了几个方法都处理不了。。
你好,不能将所有负值赋值NA,log2转化时,小于1的会变成负值,不应删除。但是需要删除的是原先TPM=0的,也就是经过log2(TPM+0.001)后= -9.965784的样本,几种方法,我习惯用的是which(data$TPM = -9.965784)得到log2(TPM+0.001)为-9.965784的行号,再删除这些行:data[-which(data$value = -9.965784),]即可
感谢进哥!
RSEM expected_count (DESeq2 standardized) (n=19,039) UCSC Toil RNA-seq Recompute
RSEM expected_count output normalized using DESeq2
unit:log2(expected_count-deseq2+1)
您好,请问用上述文件提取自己想要的癌种进行差异分析适合用什么方法,这个数据不是原始的raw count用DEseq2似乎不合适了,用limma(因为文件已经用DEseq2标准化了,所以我直接用limma进行差异分析,没有用limma标准化,总之就是感觉不太对)分析的上调基因有6000多个
你好,直接用limma即可,请问具体是哪一个癌症类型 我看看我的分析结果
您好,首先感谢解答!我分析的是胰腺癌,不过只提取了其中胰腺导管腺癌的150个样本,总的183个是包括了其他的胰腺肿瘤所以被我删除了一部分,我做差异分析之前用boxplot看了一下数据分布,确实是肿瘤组的数据看着位置高一些,我不太清楚TCGA和GTEx的差异到底是肿瘤组织和正常组织之间的差异还是因为批次效应产生的差异,因为其他人做的差异基因数目并没有这么多。筛选条件是logFC=2,P.Value = 0.05,而且我用来做差异分析的基因不是全部基因,是进行了ID转换的功能为protein-coding的基因,有19590个。用table函数看的上调下调的基因数目如下:
down stable up
240 12390 6960
(我用来做差异分析的代码,是从网络上获取的)#limma差异分析
boxplot(exp,outline=FALSE, notch=T,col=group_list, las=2)
library(limma)
design=model.matrix(~group_list)
fit=lmFit(exp,design)
fit=eBayes(fit)
deg=topTable(fit,coef=2,number = Inf)
write.table(deg, file = “deg_all.txt”,sep = “\t”,row.names = T,col.names = NA,quote = F)
logFC=2
P.Value = 0.05
k1 = (deg$P.Value < P.Value)&(deg$logFC < -logFC)
k2 = (deg$P.Value logFC)
deg$change = ifelse(k1,”down”,ifelse(k2,”up”,”stable”))
table(deg$change)
再次感谢解答!
请问您以下文件有办法分割吗?一般的电脑的受不了这么大的内存,据说可以用R的SOAR包
cohort: TCGA TARGET GTEx
gene expression RNAseq
RSEM expected_count (n=19,109) UCSC Toil RNA-seq Recompute
我没有下载这个,确实太大了 后面有时间在处理
进哥,我想问一下TCGA中count数据能和GTEx中expected count 数据合并吗,我将二者数据都进行ceiling(2^(数据)-1)处理后合并,之后用了DESeq2进行差异分析,请问这样可以吗?
应该不合适吧 count都需要原始的
进哥,我想请教一下,为什么我跑出来的泛癌分析基本上每个癌症中都有显著性差异?这和我想得到的结果是相违背的,我在一些在线网站上或者单个癌症分析都是没有那么多差异的。想请教一下怎么解决这些问题呢?
你好 和其它工具得到的结果不一致可能是统计方法的差异,比如GEPIA用的limma和one-way ANOVA,这个里面用的T-test 你可以选择其他统计方法 比如秩和检验wilcox,亦或是limma
进哥,我尝试将检验方法改了,但是还是不行。我发现我的图中有大量的负值,我觉得这可能是影响差异结果的地方。我想请教一下如何处理这些负值
您好,我想请教一下一般差异分析不是都用count文件好一些?那用TPM等标准化后的差异分析被人认可吗?GTEx数据库的可以转化为counts类型文件吗?如果可以的话,我可以向您请教如何转化为counts文件吗?
您好 从count出发去分析肯定更准确,当然count分析首先也得标准化,而TPM是标准化之后的,也可直接用于差异分析 结果当然认可。已经标准化之后的数据很难再变回count
非常感谢,谢谢您解答我的疑惑。
王老师,您好,FMR1基因跑出来没有LAML的癌症数据,并且MESO缺少正常对照的数据
进哥您好,最近一直被这个TCGA和GTEx连个数据库联合分析的事困扰,发现您的这个网站万分激动,叨扰想请教你一个问题:
1.UCSC数据库下载的CGA和GTEx样本counts数据能否直接合并了进行差异分析?
2.后续分析是必须单独下载FPKM或者TPM值进行合并后分析?合并的时候有必要再消除批次效应嘛?
感谢进哥,期待您的解答!
您好 我提供的数据是可以直接合并的,他都是log(TPM+0.001)
如果你要自己下载其它标准化数据进行合并分析 需要保证标准化方式一样,比如上述log(TPM+0.001) 批次效应可以不用 不放心可以去除一下
您说的这个是fpkm等经过标准化后的数据,在保证相同的处理方式下(如log2(counts)+1),就能直接合并分析,不用进行批次效应消除嘛?
那counts数据能不能直接合并,然后进行差异分析呢?
特别感激进哥的恢复,谢谢啦
GTEx和TCGA测序可以不用批次校准是因为他们都是NIH的项目,所使用的的平台都一样,当然肯定不是一起上机的 ,就算TCGA样本也是慢慢积累起来的。基于同一机构 同一测序平台 可以不用去除批效应 当然不放心可以检验一下
Count数据当然可以合并 然后在进行差异分析
收到,感谢进哥解答
麻烦王博帮我看下我的图,做出来好奇怪啊
可以的 加我微信 图奇怪是因为参数没有修改
进哥您好 感谢您的代码 十分好用 但是我在跑自己感兴趣的基因的时候发现无论我跑什么基因最后生成的图片表达量的显示范围都是-10到10这个区间 但是我有好几个基因的数据明显是超出这个范围的 所以柱子就会残缺 请问是否能够解决呢?感谢!
加我微信吧 数据超出范围修改y轴范围就好
感谢!
师兄你好,请问log2(TPM+1)和log2(TPM+1)的数据用来分析有没有区别?有什么办法可以互相转换呢
是log(TPM+1)和log2(TPM+0.001)
都可以的,因为表达数据一般呈现偏态分布,将其log后数据正态,这样分析更准确。因为存在表达为0 的数据,无法取对数,故在此基础加上一个数 可以是1 也可0.1,0.001,对结果影响不大
要转化的话就是幂运算变成原始TPM 再重新取对数
进哥你好,为什么我用你分割好的数据进行差异分析时用Wilcoxon秩和检验的FDR值在0.7-0.9左右?
你好,那数据看起来差异大吗?或者FDR和P值差的大吗?你发我数据看看也行 或者直接回复那个基因名称 我看看
博主您好,请问有GTEX样本的临床信息(年龄)吗?我在xena的网站没有找到
您好,我也查了一下,好像是没有年龄信息的,只有性别信息
有年龄信息的,只不过只提供了年龄区间,可以在GETx官网的metadata上找到
Error in fread(“TCGA-STAD.GDC_phenotype.tsv”, header = T, sep = “\t”, :
这个咋弄
加我微信吧,报错具体什么信息?
您好,我用您发的tpm想做cibersort,我把它(2^tpm-0.001)变成原始tpm,但是还是有负数,cibersort矩阵不能有复数,我也试了log2(TPM+1),还有负数,我不知道该咋处理了,请问我应该怎么处理这个数据呢
您好,按道理TPM不应该有负数,但是为什么逆运算之后有负值?
因为修约的原因,原先TPM有0,但是0不能取对数,因此在原先矩阵都加上0.001或1,这样得到的结果其实只保留了一定的小数位数,因此,用这个修约的数值逆运算,最后值就不等于0 ,会出现负值
就拿这个log2(TPM+0.001)来说,TPM=0时,结果为–9.965784,逆运算之后为2^(-9.965784)-0.001=-1.423899e-06
So,解决方法就是将负值赋值0,或者不采纳这些数据直接删除,即NA
负值出现的没有规律,俺甚至想手动删除负值惹,师兄可以提供一下将负值赋值为0 的代码吗
+1,可以发给我吗? 1662627418@qq.com
您好,OV的合并数据和利用我分割好的数据进行合并的代码都已上传网盘:链接:https://pan.baidu.com/s/13RcJQUZziuPHpZANkrh3_w?pwd=xjaf
提取码:xjaf
进哥你好,为什么TCGA Pan-Cancer (PANCAN)分割出来的单个癌症样本数与直接下载的相同癌症的样本数不一样呢?
是的呢,我没有去具体查过,应该是因为部分样本不符合合并的质量控制,被人为剔除了,你可以差一下相关文献 我有空的时候也看一下
想请问一下tcga_RSEM_gene_tpm.gz这个文件也是在xena上下载的吗?我好像没有找到欸
还想顺便问一下如果是fpkm数据也是一样的处理吗?那fpkm和tpm数据哪个更好啊
一样处理 保证两个数据都是FPKM
另外一边现在普遍使用TPM FPKM已经被主流抛弃了 很少用了
您好,您发的百度网盘里正常和肿瘤数据都是tpm+0.001吗,可以直接合并。之前我在网上看到差异分析需要counts,请问tpm可以吗,需要用limma包?
您好,网盘里面的可以直接合并,差异分析用limma可以,count数据用deseq2分析,当然标准化之后也可以选择其他包
是的,点击文章中的文件名 有到xena的下载链接
进哥,请问这个文件怎么下载呢?“gencode.v23.annotation.gene.probemap”
这个是从Genecode官网下载的,https://www.gencodegenes.org/human/,目前已有更新,这个v23比较老的
实际上文中文件有下载链接的
进哥你好,问下TCGA中没有正常样本,用GTEx中作对照,然后你发的这个代码是选取感兴趣基因,能发下筛选总的差异基因的代码吗。谢谢!
你好,你会一点R语言或者用过R语言进行差异分析的话,这个无非就是将两个数据集矩阵进行merge,然后分组信息就是GTEx是normal,TCGA是Tumor,按照常规方法进行差异分析。
具体差异分析方法可以选用limma包
可以自己先试试,搞不定再交流
进哥这个需要去除批次效应吗
不需要的,都是同一平台 同一机构的数据 标准化方式一样即可
师兄你好,请问将tcga和gtex数据集矩阵合并后进行差异分析,数据格式需要为counts吗?
你好 你有count数据就用count,然后用deseq2差异分析 本文提供的是log2(TPM+0.001),标准化之后的数据,直接用limma包进行差异分析即可
好滴 谢谢师兄~
进哥您好,现在的XENA数据库是有已经合并好的TCGA+GTEx数据,我做了TCGA与GTEx样本之间表达量的分析,发现不管是PCA还是箱线图两个组都有比较明显的差别,所以我感觉似乎数据并没有做去除批次效应处理,但按照您的说法,他们的数据不用去除批次效应,那在做差异分析前是不是还需要标准化处理?如果需要,请问这种不同组之间的标准化用R语言要怎么实现呢?我知道的normalizeBetweenArrays函数只适用于同组之间的处理。
您好 可能主要原因在于样本不一样 GTEx主要正常组织,TCGA主要癌症 所以PCA/箱线图肯定分开 normalizeBetweenArrays适用于芯片数据,测序的话不太清楚
进哥你好,我想咨询一下做弥漫大b淋巴瘤,GTEx里应该提取那个组织的表达组数据啊,应该是淋巴结吧,我没有找到。。
额,这种血液肿瘤就直接以blood做对照吧,最好是以同种组织的正常组织作为对照,没有的情况下找相近的
进哥哥,您最后用了t.test,可以认为tcga和gtex两组间是满足正态方差齐吗?
您好,终于有人发现了这个统计上的漏洞。事实上,大部分癌症类型是满足的,对于部分样本少,或者有缺失数据的可能就不满足正态方差齐,这时候用Wilcoxon秩和检验更加合适,选择t.test只是图个方便。t检验和Wilcoxon秩和检验
您好,之前看过有人说TPM或者log2TPM的数据只能做非参数的秩和检验。例如:
x <- c(100,200,300)
y <- c(1000,2600,3000)
t.test(x,y) # p=.8787t.
test(log(x),log(y)) # p=.071
wilcox.test(x,y) # p=0.1
wilcox.test(log(x),log(y)) # p=0.1
在这样的数据变换中,t检验的p值会改变,但是wilcox.test不会。
不知道有无道理
博主您好,我在把YTHDC2换了一个目的基因,出的图会变形,没办法调整Y轴
您好,你指的变形是样本量的位置是吗?这一句调整位置,改一下y=:geom_text(data=count_N, aes(label=n, y=-9,color=Group)
如果其它问题可以详细描述一下 或者加我微信
进哥好,请问下target是否可以一次多个,进行分析比较
你想要一次分析几个基因在33种癌症中的表达吗?要做当然可以,关键看你最终将以什么形式呈现,可以加微信讨论
gtex_exp <- fread("gtex_RSEM_gene_tpm.gz",data.table = F)文件过大,无法打开,更改data.table=T勉强可以,想问进哥哥有啥办法只提取这个大数据里我需要的数据即可,不必完全打开。
你好 之前评论回复里面有分割的GTEx和Pancancer数据,你可以只下载你需要的组织类型数据,然后进行合并分析,如果还是需要多种癌症,写个循环即可
是这样的,我用的gz文件和进哥哥展示的还不太一样,我使用的是transcrip expression的,所以我是不是得自己分割了,,,,
进哥,我还有一个问题,THYM正常对照可以用blood吗?虽然胸腺里面是免疫细胞,但是和全血还是差别很大的
您好 这个我不清楚 讲道理应该不可以,你看文献怎么处理的,如果实在没有正常组织,可以考虑分析不同分期的差异
tcga<-tcga[,c(3,2,6,5)]
Error in `[.data.frame`(tcga, , c(3, 2, 6, 5)) : 选择了未定义的列
进哥,我按照你的代码整理tcga表型文件的时候提取3,2,6,5例时显示有一例不存在,我看了tcga table只有5列,detail ,TCGA, sample, sample_type_id, sample_type.这该怎么解决?
tcga$type <- ifelse(tcga$sample_type=='Solid Tissue Normal', paste(tcga$TCGA,"normal_TCGA",sep="_"),paste(tcga$TCGA,"tumor_TCGA",sep="_")) 这一行运行了吗 还是运行报错了?因为你没有type这一列
进哥,像UVM这种联合GTEX都拿不到正常组织数据的癌种,怎么分析基因差异呢?
额 确实没有,我也无能为力,不过可以考虑分析不同分期的差异
请问您的TCGAphenotype文件是什么样的来源?是进行过处理的吗?还是从xena直接下载的呢?如果进行过处理请问是怎么样处理的呢?
您好 这个文件是xena直接下载的,保存的是癌组织来源,没有进行处理,直接与表达数据合并
张博士,你好,请问下DLBCL中GTEx的数据用的是全血细胞吗?可以把DLBCL的TCGA数据和GTEx数据发我吗?
您好,我姓王,前面的回复里面有所有分割好的数据,链接:https://pan.baidu.com/s/17blyTZb-Kni8u9yqIsweyA?pwd=eqsp
提取码:eqsp
您好,请问可以把GTEx的数据和GEO的数据合并分析吗?需要下载什么样的数据?研究的病理亚型在TCGA 样本量太少了
合并的话就是保证使用的TCGA和GTEx数据集是同一种标准化方式,比如这篇文章里用的是log2(TPM+0.001)。
但是您说所研究的病理亚型在TCGA样本量太少,那你合并GTEx也没有帮助哇,GTEx都是正常组织。如果TCGA无法满足,可以尝试搜索相关GEO数据库或一些其他肿瘤数据库,比如ICGC等
您好,进哥。我想咨询一下,您的这些数据有正常脑组织样本数据吗?与 MiRNA 有关的。期待并感谢您的解答
正常脑组织数据有的,但是GTEx数据库只有genotype和gene expression数据,应该是没有mirna expression数据
您好!想要您 “tcga_RSEM_gene_tpm.gz ”这个数据,因为文件很大,下载不下来,非常感谢!
你好,我这边有下载好的,可是一样很大,网盘发你下载也会很慢很慢。另外我有根据组织类型分割好的,前面评论有链接
进哥,如果方便,恳请发一份卵巢癌及子宫内膜癌的TCGA+GTEx整合的表达矩阵文件给我吧,谢谢!
好的,你是已经加我微信了吗?我发你代码,授人以渔
谢谢您!想问一下分割好的数据,即来自TCGA和GTEx两个都是log2(TPM)后的吗?
log2(TPM+0.001) 具体可以查看xena说明
博主你好,求一份卵巢癌TCGA+GTEx整合的表达矩阵文件,谢谢!
好的 晚一点发你
上一个留言中有Pancancer和GTEx分割好的数据,merge一下就可以整合
我跑上面的代码出现这一行错误:
> write.table(exp,”Merge gene expression/YTHDC2 expression.txt”,row.names = F,quote=F,sep=’\t’)
Error in file(file, ifelse(append, “a”, “w”)) :
cannot open the connection
In addition: Warning message:
In file(file, ifelse(append, “a”, “w”)) :
cannot open file ‘Merge gene expression/YTHDC2 expression.txt’: No such file or directory
因为你路径下不存在”Merge gene expression/YTHDC2 expression.txt”,你需要改成你自己电脑上保存的路径及文件名
把YTHDC2换了一个目的基因,出的图会变形,需要调整哪个参数?
具体是怎样的变形 要不你加我微信 讨论 18021308280
您好,同求一份卵巢癌的TCGA(379份)和GTEX(88份)合并后的数据,另外我已经对这两个数据处理完了,但是它们的表达量值相差较大,请问应该如何合并呢
您好,OV的合并数据和利用我分割好的数据进行合并的代码都已上传网盘:链接:https://pan.baidu.com/s/13RcJQUZziuPHpZANkrh3_w?pwd=xjaf
提取码:xjaf
博主,我可以求一个卵巢相关的GTEx的数据吗,GTEx的数据我试了很多种方法都下不了。非常感谢!!!!!!
OK,发您邮箱,注意,这个数据是log2(TPM+0.001)
谢谢!!!!!!!!!
我也求一份!进哥
想求问有没有只计算GETx某基因表达量的代码呀
你好 没有代码,这个比较简单 直接利用GTEx画图,分组柱状图 此外,GETx网站提供了表达量分析,你看看GTEx:基因型和基因表达量关联数据库
举个例子,你点进去看
博主您好,请问我想要看看Xp上的某些基因在不同组织中XCI ratio,要怎么做啊,谢谢
这个我没有涉及过,您加我微信 探讨一下
有针对单个癌的联合吗
可以呀,把泛癌和GTEX数据拆分成单个癌症的就可以啦
之前读取数据的时候内存不够,所以我用了memory.limit(newLimit)修改了内存,但是现想把这个再降回来,现在R语言 用了太多内存。怎么给清空呢。
used (Mb) gc trigger (Mb) max used (Mb)
Ncells 6880145 367.5 11096502 592.7 11096502 592.7
Vcells 14379954 109.8 409974109 3127.9 903763272 6895.2
请问一下,单个基因进行差异分析,tpm数据需要经过处理吗,有些是负值。
你好 这里面用的数据是经过log2(tpm+0.001)处理的,对于有些表达量低的,转化之后是负数。所以 这些数据不需要进行其他处理,直接可以使用
如果希望将log2(tpm+0.001)的数据进行转换为log2(tpm+1),应该用什么代码呢?尝试了很久都不行,希望能得到博主的帮助
咦 反向2^value-0.001得到原始TPM 在进行log不行吗?问题出在哪?不行直接加微信讨论
进哥GTEx与TCGA连用的时候是不是去除批效应,GEPIA那个网站去除了吗?
你好,事实上GEPIA这个工具使用的也是从XENA数据库中经过标准化的数据:UCSC Xena project recomputed the TCGA and GTEx raw RNA-Seq data using a standard pipeline, which makes two datasets compatible. As a result, the TCGA and GTEx data could be integrated for very comprehensive expression analysis. 所以我认为利用Xena数据库的数据分析时不需要再另外进行去批效应处理
博主您好,我下不了GTEx的数据,请问您可以提供下吗,谢谢
Xena数据库国内访问没应该有问题,是下载太慢吗?我有下载好的,但是一样很大,放网盘再给你下载一样也很慢,我这边也有按照肿瘤类型分割好的数据,如果不做泛癌我可以发你需要的癌症数据
我需要BRCA(乳腺癌)跟THCA(甲状腺癌)两个类型的数据,可以发我邮箱吗?谢谢
你好,是只需要pancancer的数据吗?还是GTex的。需要等周一回学校,在学校电脑
我需要TCGA+GTex联合的数据,根据肿瘤类型分割好的数据,就只要BRCA跟THCA 这两个瘤种数据
链接:https://pan.baidu.com/s/17blyTZb-Kni8u9yqIsweyA?pwd=eqsp
提取码:eqsp
在家里电脑又分割了一下,我把GTEX和pancancer的所有分割好的数据发你,你自己下载下来合并分析,在R里面用merge根据Ensemble ID合并即可
王博士您好!我想请问一下计算t检验显著性时会报错Error in `mutate()`:
! Problem while computing `p = purrr::map(…)`.
Caused by error in `if (stderr < 10 * .Machine$double.eps * max(abs(mx), abs(my))) …`:
! missing value where TRUE/FALSE needed
Run `rlang::last_error()` to see where the error occurred.
是什么原因?谢谢!
您好,这个问题应该是你的表达量那一列非数值型,你查看一下基因表达量那一列是否有NA,也看一下这一列的数据类型是否为numberic,如果有,可以Excel删除这些NA值再导入R,或者在R中用as.numberic()把表达量那一列转换一下,若有问题,可以加微信讨论
好的,谢谢博士!
多谢
请问一下,我看到在XENA网站上下载的GTEx和TCGA数据的TPM计算方式不一样(GTEX是log2(TPM+0.001),TCGA是log2(TPM+1)),可以直接合并进行分析吗?不用转化成统一的格式吗?还有我看gepia网站的差异分析中是没有负值的,但是我做的TPM中是有负值的,log2转化后就是nan,该怎么办?
TCGA Pan-Cancer (PANCAN)
GTEX
你好,合并分析请使用上述pan-cancer数据集提取特定癌症进行合并,单位都是log2(TPM+0.001)
标准化方式或单位不一样都是不可以直接合并分析的
博主您好,我想问一下在GTEX中下载的这个gtex_RSEM_gene_tpm.gz 正常样本中,来自血液的样本编号是什么呢? 或者是骨髓来源的 样本编号是什么? 网上找了半天也没找到,恳请您的答复。
您好,这个样本注释信息在GTEX_phenotpye.gz 文件中有,有关于样本来源组织的信息(包括whole blood)
这个文件文中有链接下载
写的很棒,学到了很多,但是下面这两个一起跑会显示:Error: cannot allocate vector of size 523 Kb,始终没有找到解决的办法?求大神给解答一下。谢谢
gtex_exp <- fread("gtex_RSEM_gene_tpm.gz",data.table = F)
gtexS <- gtex_data[ gtex_data$sample%in%colnames(gtex_exp)[-1],]
tcga_exp <- fread("tcga_RSEM_gene_tpm.gz",data.table = F)
tcgaS <- tcga[tcga$sample %in%colnames(tcga_exp)[-1],]
网上找的教程,可以试试:
cannot allocate vector就是典型的数据太大读不了
一、升级硬件
二、改进算法
三、修改操作系统分配给R的内存上限, memory.size(T)查看已分配内存
memory.size(F)查看已使用内存
memory.limit()查看内存上限
object.size()看每个变量占多大内存。
memory.size()查看现在的work space的内存使用
memory.limit()查看系统规定的内存使用上限。
如果现在的内存上限不够用,可以通过memory.limit(newLimit)更改到一个新的上限。注意,在32位的R中,封顶上限为4G,无法在一个程序上使用超过4G (数位上限)。这种时候,可以考虑使用64位的版本。
你好,请问一下我发现内存读取不了之后使用memory的一些函数都会报下面这种错是为什么呢
Warning message:
‘memory.size()’ is no longer supported
根本就不能使用这个函数
我也不清楚 应该是新版取消了这个函数,好像现在R进程的可用内存与系统中的可用内存相同,这个函数就没有用处了
“samplepair.txt”请问师兄这个文件是哪里来的?谢谢!
同学你好,原文数据来源已添加链接
谢谢师兄,我自己试了一下,跑不出结果,哭了
1.注释第三步的时候出现报错
> tcga_exp source(“GeomSplitViolin.R”)
Error in file(filename, “r”, encoding = encoding) : 无法打开链结
此外: Warning message:
In file(filename, “r”, encoding = encoding) :
无法打开文件’GeomSplitViolin.R’: No such file or directory
这个我完全看不懂,后面几乎都报错,也有“无法配置矢量”的报错
谢谢师兄指点!
source(“GeomSplitViolin.R”)这一句多余的,原本这个文件里面是一个分裂小提琴图的函数,后面并没有用到。后面不应该会报错,你可以加我微信讨论,在我的简历里面有手机号和二维码