方法1 windows下使用SRA Toolkit下载
首先在官网下载SRA Toolkit windows版本软件。
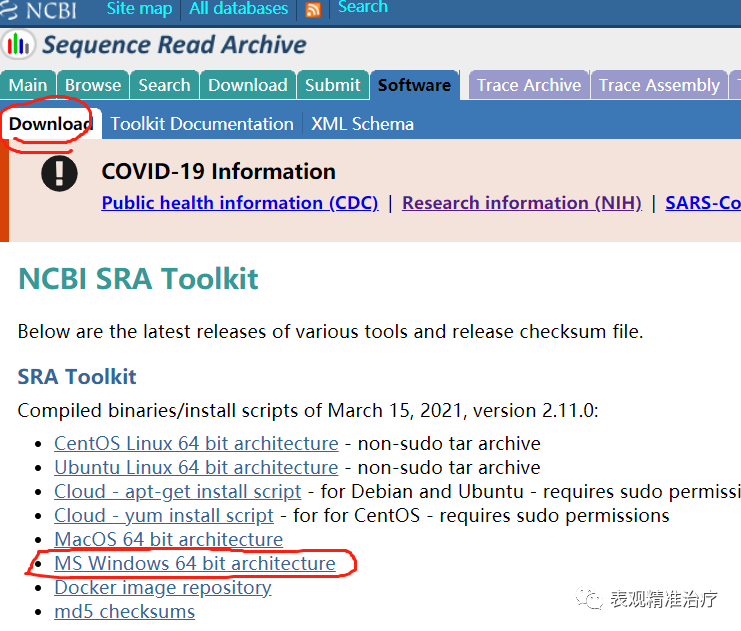
然后解压,安装。
#存储路径sratoolkit.2.11.0-win64binvdb-config --interactive进入安装界面
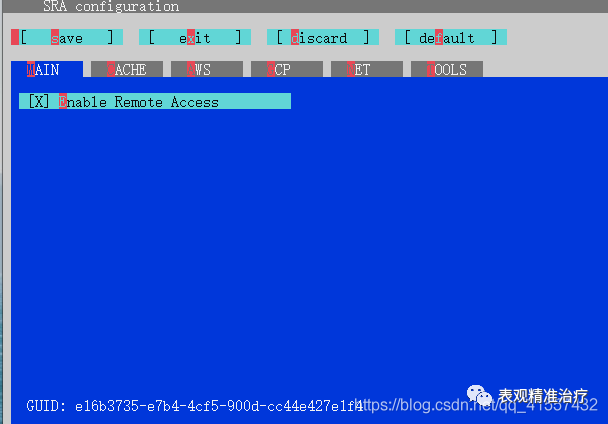
为了防止各种插件出错,保险起见,选择默认。
按上下键选择,按“s”保存,再按“exit”退出。
如图所示既是安装成功
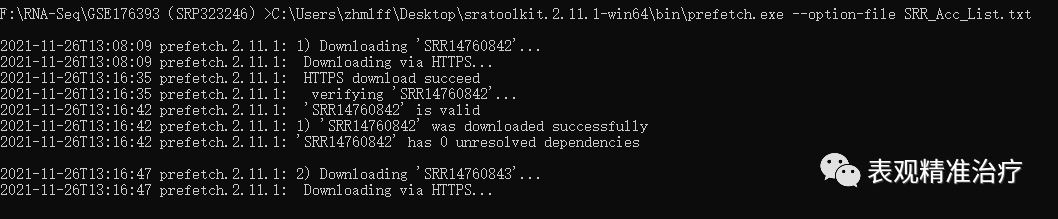
下载数据很方便,进入SRA数据库,选择要下载的数据,下载其SRR_Acc_List.txt文件,在数据存储目录中运行以下代码即可:
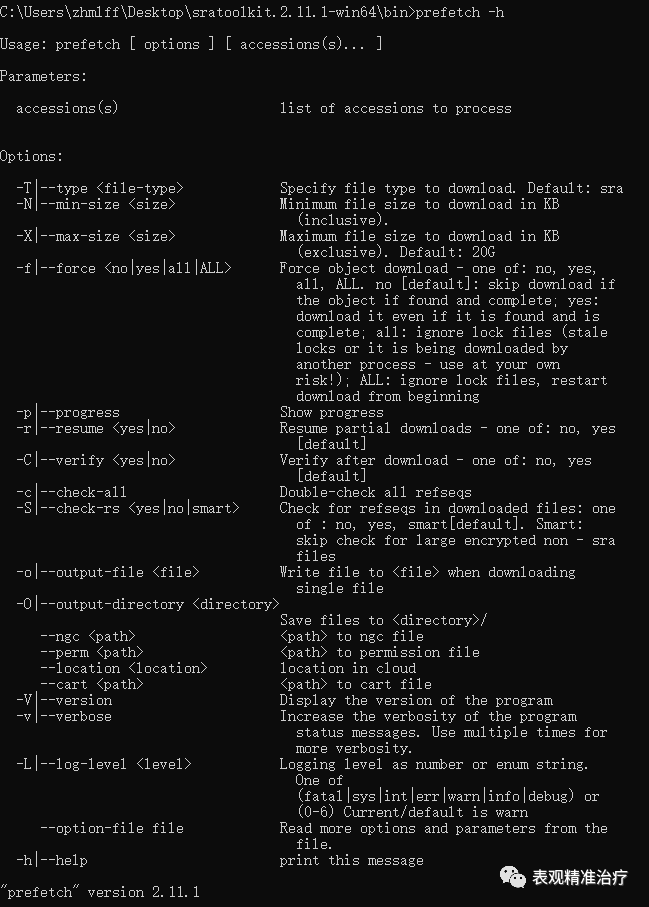
#存储路径sratoolkit.2.11.1-win64binprefetch.exe --option-file SRR_Acc_List.txt
按照以下方法可找到SRR_Acc_List.txt文件。
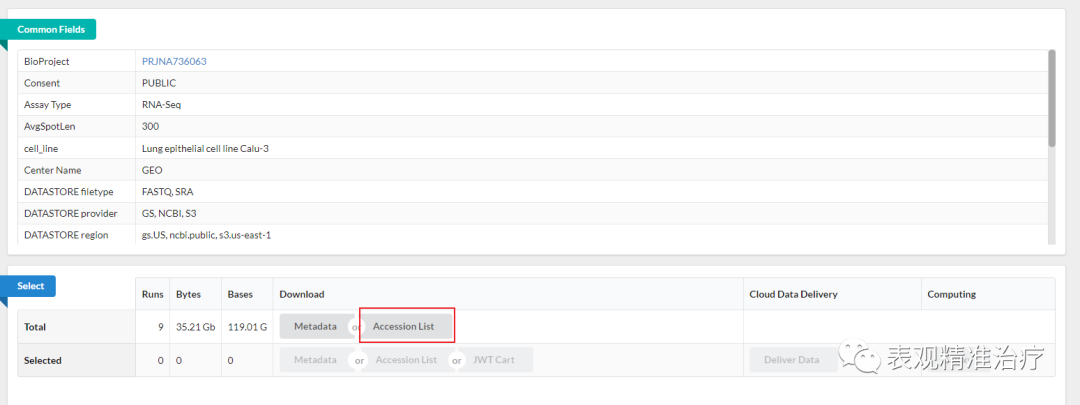
第一步 进入目标数据的GEO信息页面,点击红框位置(GEO数据库如何使用,我们随后详细介绍:辛苦大家帮我记住这个坑 )
)
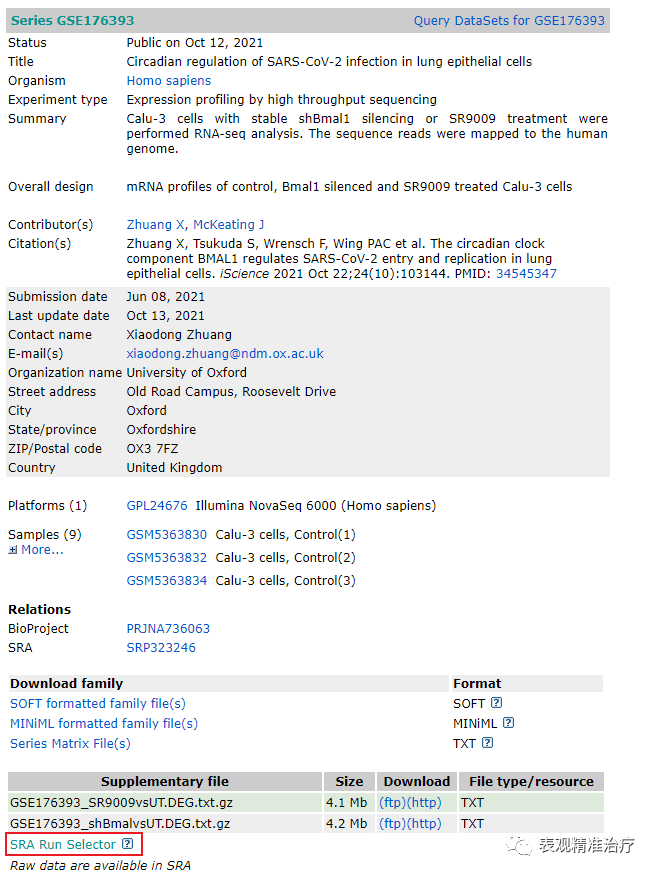
第二步 进入下图页面后点击红框位置,另存为SRR_Acc_List.txt文件至数据存储路径
开始下载数据会是这样,最后等待下载完成就好了。
方法2 使用sra-explorer下载
SRA Explorer(https://sra-explorer.info/)可以用来生成SRA数据下载命令
接着上面介绍的,选好数据后,可以找到数据编号(GSE号或SRA数据号都可以,例如上面的就是GSE176393或SRP323246)输入搜索框。操作如下图。
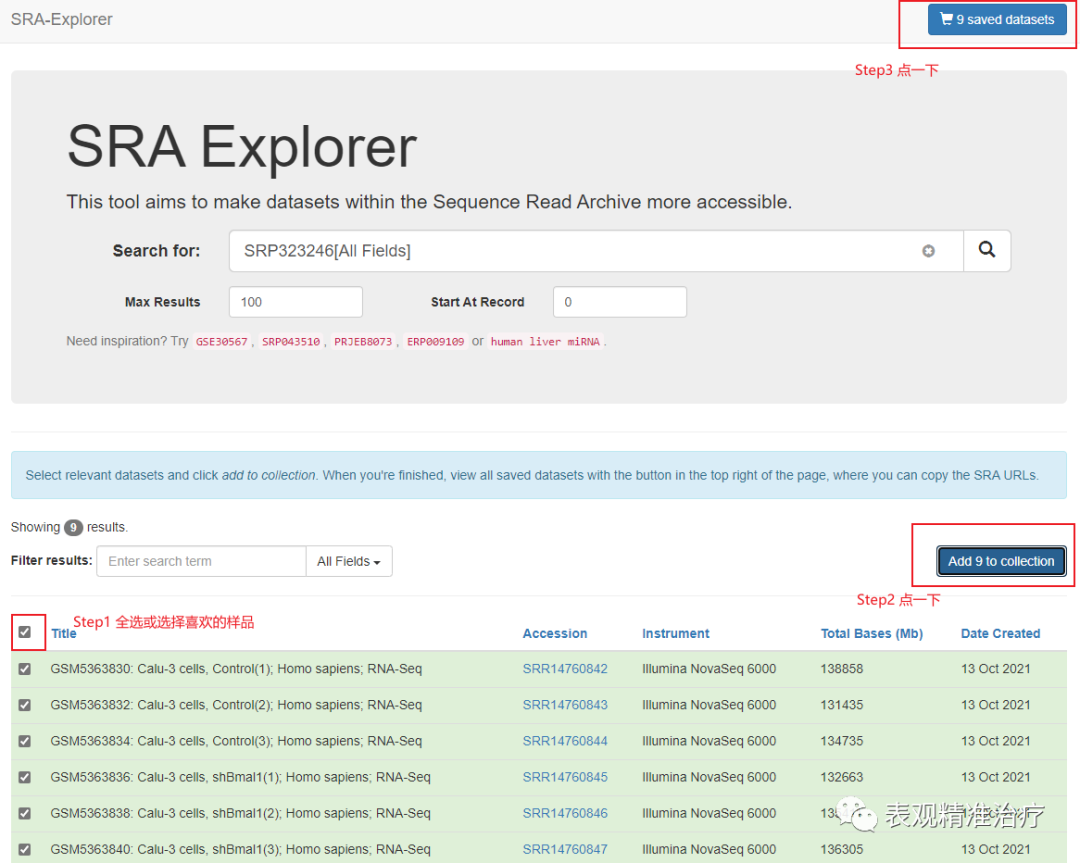
完成上述三步后会出现这个。
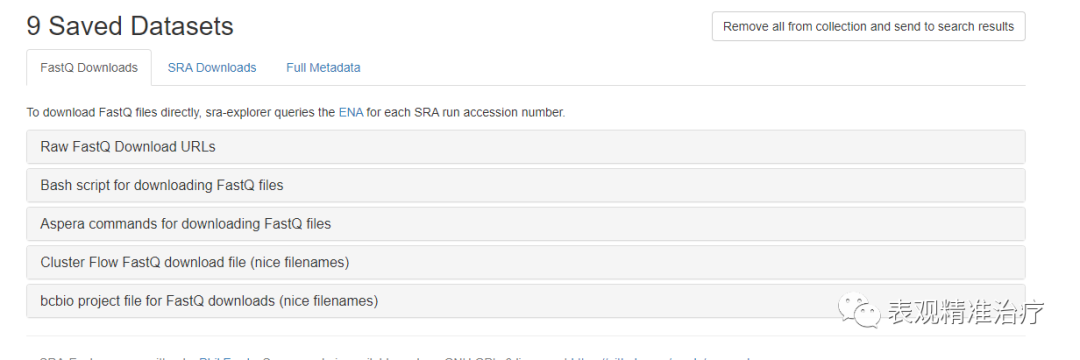
这里我们可以看到很多关于各种数据类型的URL,你可以选择直接下载FASTQ格式文件,也可以选择下载SRA文件。我选择直接下载fastq格式文件,方便操作。
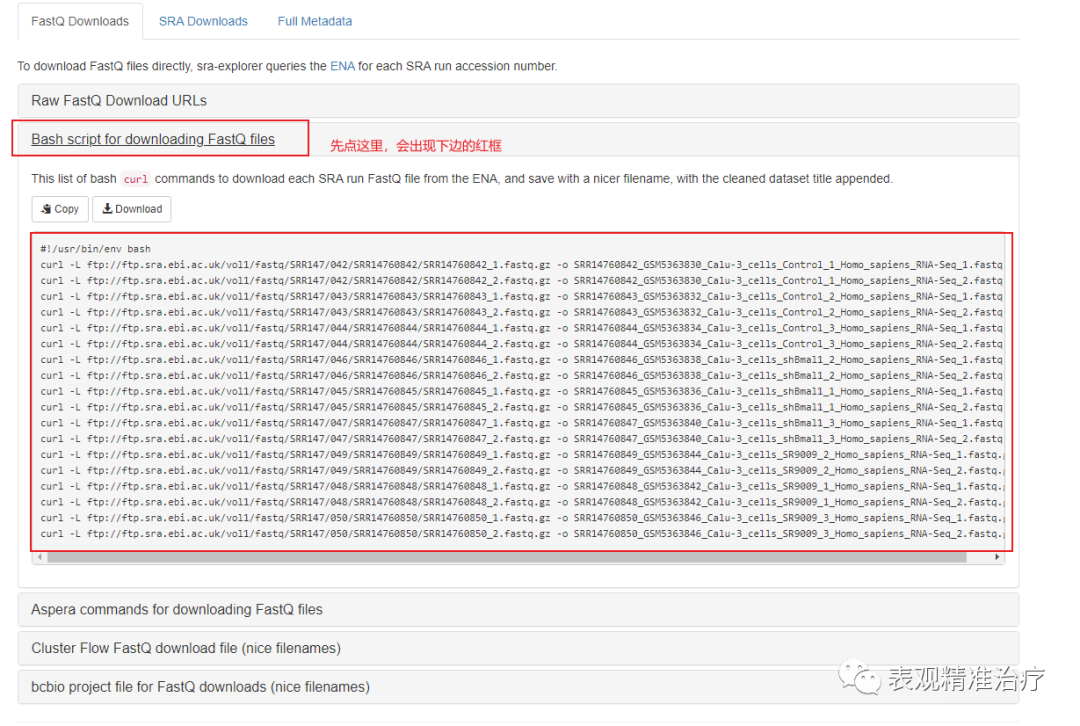
出现下载命令后有两个选择,1. 比较笨,在linux子系统中一个一个运行。2. 将命令复制进一个.sh中当做一个shell脚本批量下载。
vim download.shnohup bash download.sh & #后台远行 运行情况写入nohup.out文件中。
以上方法可根据每个人的爱好使用,只要网络环境好均可下载。
如果使用方法二下载,可直接使用进行后续分析
如果使用方法一下载,会将.sra数据存入以数据编号建立的文件夹中,需要先将数据全部整理入一个文件夹进行操作,这样会方便很多。
上代码!
##设置一个循环可以批量操作mkdir downloadcat SRR_Acc_List.txt | while read linedomv $line/$line.sra download/$line.sradone
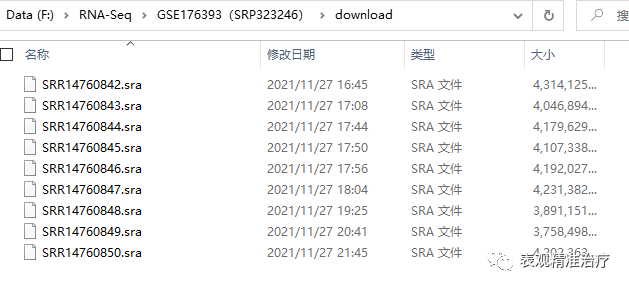
随后应用fasterq-dump将.sra数据转换为.fastq数据,
fasterq-dump需要在WSL上安装,运行以下代码即可安装:
conda install -c bioconda sra-tools=2.11.0##安装完成后先查看一下fasterq-dump的帮助信息,看看如何使用
fasterq-dump -h
Usage: fasterq-dump [ options ] [ accessions(s)... ]
Parameters:
accessions(s) list of accessions to process
Options:
-o|--outfile <path> full path of outputfile (overrides usage
of current directory and given accession)
-O|--outdir <path> path for outputfile (overrides usage of
current directory, but uses given
accession)
-b|--bufsize <size> size of file-buffer (dflt=1MB, takes
number or number and unit where unit is
one of (K|M|G) case-insensitive)
-c|--curcache <size> size of cursor-cache (dflt=10MB, takes
number or number and unit where unit is
one of (K|M|G) case-insensitive)
-m|--mem <size> memory limit for sorting (dflt=100MB,
takes number or number and unit where
unit is one of (K|M|G) case-insensitive)
-t|--temp <path> path to directory for temp. files
(dflt=current dir.)
-e|--threads <count> how many threads to use (dflt=6)
-p|--progress show progress (not possible if stdout used)
-x|--details print details of all options selected
-s|--split-spot split spots into reads
-S|--split-files write reads into different files
-3|--split-3 writes single reads into special file
--concatenate-reads writes whole spots into one file
-Z|--stdout print output to stdout
-f|--force force overwrite of existing file(s)
-N|--rowid-as-name use rowid as name (avoids using the name
column)
--skip-technical skip technical reads
--include-technical explicitly include technical reads
-P|--print-read-nr include read-number in defline
-M|--min-read-len <count> filter by sequence-lenght
--table <name> which seq-table to use in case of pacbio
--strict terminate on invalid read
-B|--bases <bases> filter output by matching against given
bases
-A|--append append to output-file, instead of
overwriting it
--ngc <path> <path> to ngc file
--perm <path> <path> to permission file
--location <location> location in cloud
--cart <path> <path> to cart file
-V|--version Display the version of the program
-v|--verbose Increase the verbosity of the program
status messages. Use multiple times for
more verbosity.
-L|--log-level <level> Logging level as number or enum string.
One of
(fatal|sys|int|err|warn|info|debug) or
(0-6) Current/default is warn
--option-file file Read more options and parameters from the
file.
-h|--help print this message
"fasterq-dump" version 2.11.0
通过help文档可以确定,我们的双端测序,所以需要把文件分成两个,故设置参数–split-files;由于fasterq-dump不能直接生成.gz压缩文件,所以后续还需手动压缩节省分析数据所用的空间。
mkdir rawdata #建立一个存储数据的文件夹
##结合目的选择好参数,开始批量转换
cat SRR_Acc_List.txt | while read line
do
fasterq-dump -e 12 --split-files download/$line.sra -O rawdata
done

gzip *.fastq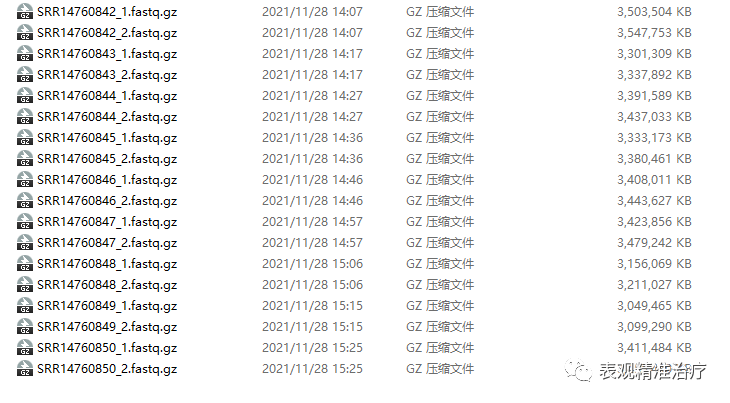
得到这样的数据就可以很方便地进行下面的分析啦。
博主好~我想请教一下 在使用第一种方法下载SRR文件的时候 出现了 ‘SRR18134878’ has 502 unresolved dependencies是为什么呀 怎么解决呢 谢谢~