此程序用来下载TCGA中的FPKM数据,TPM转化,以及mRNA与lncRNA提取,以便进行分析。另外进行差异分析需要count数据,只需要把workflow_type <- “HTSeq – FPKM”中的FPKM换成count。
如有问题请联系进哥哥(代码中提取lncRNA或mRNA时有两个参考文件联系进哥哥获取)
############################################################
# Producer=Jin Wang
###########################################################
# 安装TCGAbiolinks包
#source("https://bioconductor.org/biocLite.R")
#biocLite("TCGAbiolinks")
###########################################################
# 加载需要的包
library(SummarizedExperiment)
library(TCGAbiolinks)
###########################################################
# GDC: https://portal.gdc.cancer.gov/
###########################################################
# 设置环境参数
work_dir <- "D:\study\TCGA\GDCdata/lnc_mRNA"
# 设置程序参数,下载count
project <- "TCGA-LUAD"
data_category <- "Transcriptome Profiling"
data_type <- "Gene Expression Quantification"
workflow_type <- "HTSeq - FPKM"
legacy <- FALSE
# 设置工作目录
setwd(work_dir)
# 下载基因表达量,count/FPKM数格式的结果
DataDirectory <- paste0(work_dir,"/GDC/",gsub("-","_",project))
FileNameData <- paste0(DataDirectory, "_","RNAseq_HTSeq_FPKM",".rda")
# 查询可以下载的数据
query <- GDCquery(project = project,
data.category = data_category,
data.type = data_type,
workflow.type = workflow_type,
legacy = legacy)
# 该癌症总样品数量
samplesDown <- getResults(query,cols=c("cases"))
cat("Total sample to download:", length(samplesDown))
# TP 样品数量
dataSmTP <- TCGAquery_SampleTypes(barcode = samplesDown, typesample = "TP")
cat("Total TP samples to down:", length(dataSmTP))
# NT 样本数量
dataSmNT <- TCGAquery_SampleTypes(barcode = samplesDown,typesample = "NT")
cat("Total NT samples to down:", length(dataSmNT))
# 下载数据, 数据比较大,耐心等待
GDCdownload(query = query,
directory = DataDirectory,files.per.chunk=6,method = "api")
# 保存结果,方便后面使用
data <- GDCprepare(query = query,
save = TRUE,
directory = DataDirectory,
save.filename = FileNameData)
# 表达量提取
data_expr <-as.data.frame(assay(data))
dim(data_expr)
#fpkmToTpm
fpkmToTpm <- function(fpkm)
{
exp(log(fpkm) - log(sum(fpkm)) + log(1e6))
}
data_expr_Tpm<- as.data.frame (apply(data_expr , 2, fpkmToTpm))
expr_file_F <- paste0(DataDirectory, "_","All_HTSeq_FPKM",".txt")
expr_file_T <- paste0(DataDirectory, "_","All_HTSeq_TPM",".txt")
write.table(data_expr, file = expr_file_F, sep=" ", row.names =T, quote = F)
write.table(data_expr_Tpm, file = expr_file_T, sep=" ", row.names =T, quote = F)
# 提取Gene 表达量矩阵
gene_info = read.table("./ref/gene_info.txt",header = 1)
cat("Total Gene annotation:", dim(gene_info)[1])
gene_selected <- row.names(data_expr_Tpm) %in% gene_info$Gene_id
gene_expr <- data_expr_Tpm[gene_selected,]
cat("Total Gene Selected:", dim(gene_expr)[1])
gene_expr_file <- paste0(DataDirectory, "_","Gene_HTSeq_TPM",".txt")
write.table(gene_expr, file = gene_expr_file, sep=" ", row.names =T, quote = F)
# 提取lncRNA 表达量矩阵
lncRNA_info = read.table("./ref/lncRNA_info.txt",header = 1)
cat("Total LncRNA annotation:", dim(lncRNA_info)[1])
lncRNA_selected <- row.names(data_expr_Tpm) %in% lncRNA_info$Gene_id
lncRNA_expr <- data_expr_Tpm[lncRNA_selected,]
cat("Total LncRNA Selected:", dim(lncRNA_expr)[1])
lnc_expr_file <- paste0(DataDirectory, "_","LncRNA_HTSeq_TPM",".txt")
write.table(lncRNA_expr, file = lnc_expr_file, sep=" ", row.names =T, quote = F)
如需下载其它数据,比如miRNA,拷贝数等等,对应修改参数即可。
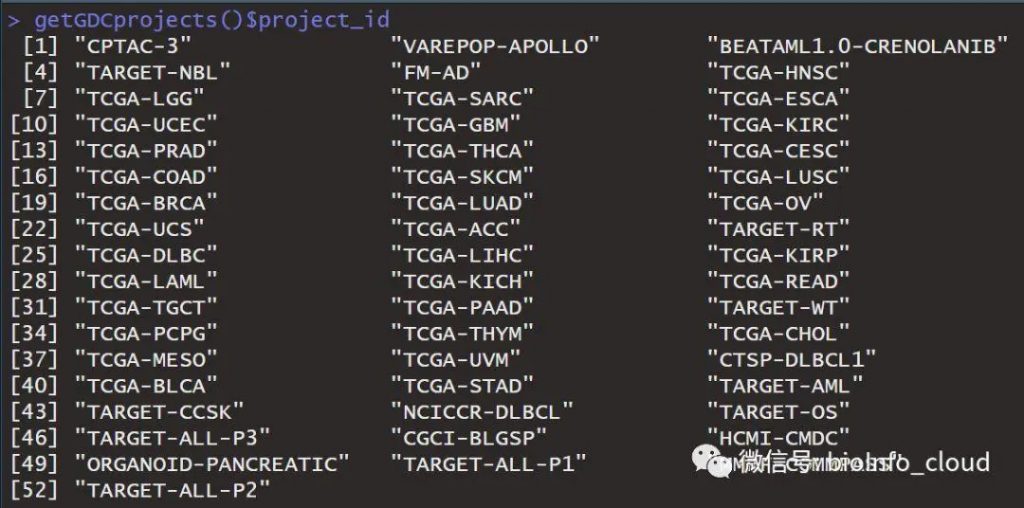
project:
可以使用getGDCprojects()$project_id得到各个癌种的项目id,总共有52个ID值。
workflow.type:
HTSeq – FPKM-UQ:FPKM上四分位数标准化值
HTSeq – FPKM:FPKM值/表达量值
HTSeq – Counts:原始count数
STAR – Counts
后续分析:
楼主你好 你这个 R 的版本是多少
R版本没啥影响,我是4.3.1
王老师您好!在使用您“TCGA_download_clinical.r”中的代码:
# 整合下载好的数据
clinical clinical <- GDCprepare_clinic(query, clinical.info = "patient",directory = DataDirectory)
|================ | 24%Error in read_xml.character(xmlfile) :
Start tag expected, '<' not found [4]
可以麻烦王老师看看怎么解决吗?万分感谢!
您好,这个的原因是,临床信息非常的复杂,有不同格式的文件,而我们需要下载的patient对应的格式只有XML格式,所以需要在GDCquery 时指定file.type=”xml” 即可。
project <- "TCGA-COAD" data_category <- "Clinical" data_type <- "Clinical Supplement" file_type = "xml" legacy <- FALSE query <- GDCquery(project = project, data.category = data_category, data.type = data_type, file.type = file_type, legacy = legacy) 顺便说一句,临床信息的数据我通常在Xena上下载,上面都是整理好的,比这个方便
你好,在运行代码保存数据这步时,会出现显示应用程序错误,然后就中止运行重启了,请问是什么原因?该如何解决?求答,谢谢!
是保存最终的结果吗?write.table(gene_expr, file = gene_expr_file, sep=” “, row.names =T, quote = F)?
这个不应该会出错,要不试试write.csv看看
进哥哥,是不是不可以同时下载2个癌症的数据,我设置project后,后面运行到GDCdownload时一直反应path错误,看来是path不能设置两个。
你好,我没有试过,不过按照你说的确实不行,不过你可以把下载的函数包成函数,再用循环同时下载多个
进哥哥想问一下那两个txt文件是不是肺癌的数据,如果我做的事胃癌方向的怎么找胃癌的txt文件
你好,那是针对所有的基因,通用的基因注释文件
进哥哥,为什么这个代码得到的lncRNA和我平常在网上看到的表达矩阵不一样
你指的不一样是指什么不一样,数值大小?这是FPKM标准化的,一般使用的是TPM,可以用公式转换,还是其它方面不一样?
进哥哥,您好!上面您提供的两个参考文件链接失效了,请问可以再次提供一份吗?谢谢!
你好,链接没有实效哇:链接:https://pan.baidu.com/s/1NV3-8EW7zkZs2ne7lMvVqg
提取码:sxbn
另外,也发你邮件了
进哥哥您好,我最近在学习生信分析,很高兴能刷您网站上的学习资料,请问是否方便发我您代码中提取lncRNA或mRNA时的两个参考文件。非常感谢!
您好,lncRNA.info怎么获取的呢?可以用相同的方法获取miRNA的id吗?
你好 前面给别人的回复中有提供网盘链接。miRNA数据下载下来之后里面是miRbase中的Accession number,可以下载下来之后进行转换成我们常见形式
棒!谢谢分享.
您好,请问gene_info.txt和lncRNA_info.txt两个文件是怎么下载的呢?谢谢!
发你邮件了 这边也发你链接
链接:https://pan.baidu.com/s/1NV3-8EW7zkZs2ne7lMvVqg
提取码:sxbn
你好,进哥哥,你的俩个TXT是什么下载的呢?不知道能不能发给我一下,感谢。
回复你邮件喽
您好,能否分享代码中提取lncRNA或mRNA时有两个参考文件。
已邮件发你了
您好,gene_info.txt和lncRNA_info.txt文件是怎么下载的
发你邮件了
老师 我也需要这个
咦?下载链接没有失效哇,前面评论里有,链接:https://pan.baidu.com/s/1NV3-8EW7zkZs2ne7lMvVqg
提取码:sxbn
进哥哥您好。我是南方医科大学临床医学专业的一名大三的学生,目前在跟随广州市南方医院的老师做lncRNA相关的研究。看见您的代码,感觉很有收获。请问可以麻烦您将代码中两个参考文件(提取mRNA和lncRNA(发给我吗?
不胜感激!
好的 我发你邮箱了 欢迎一起学习交流
您好,我也想要提取lncrna的文件,同求。
邮件给你了
链接:https://pan.baidu.com/s/1NV3-8EW7zkZs2ne7lMvVqg
提取码:sxbn