在做完m6A测序后,如何对差异甲基化的基因进行验证。简单来说就是IP后如何进行qPCR 实验。我们遇到的第一个疑问就是究竟能用多少RNA来做后期验证呢?换句话说,假如要验证10个基因,究竟需要多少total RNA或polyA的RNA才合适呢?
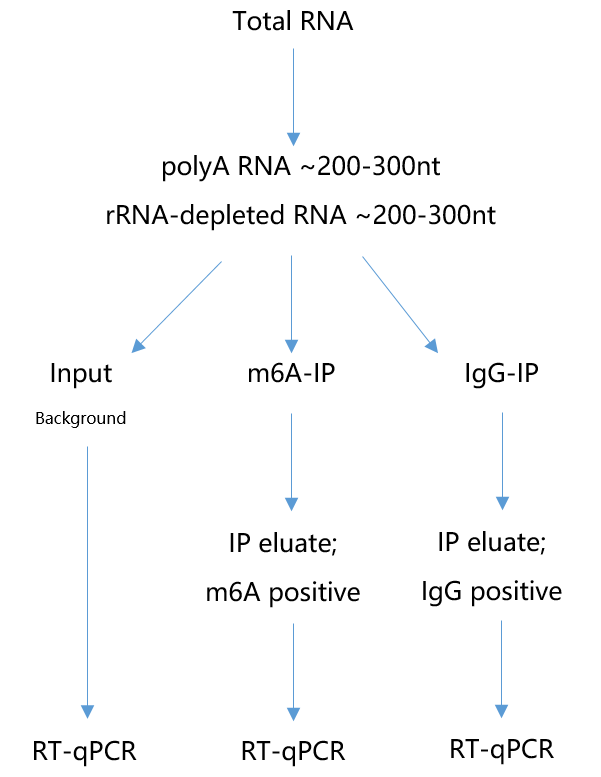
所以到这里我们的实验部分已经完成了,简单总结下就是想要做够10个左右基因的验证,必须至少提供>200ug的total RNA以上。富集的RNA类型根据自己要求出发,如polyA RNA或rRNA-depleted RNA等,最后就是打断成200-300nt左右的长度。用于qPCR的RNA一共分成3类,即input、m6A-IP和IgG-IP,其中input占m6A-IP起始量约10%。如果用数字来表示那就是input需要100ng,m6A在IP前需要1ug,IgG在IP前也需要1ug。
所以接下来我们就要详细讨论下m6A-IP-qPCR 是如何计算相对表达量的。
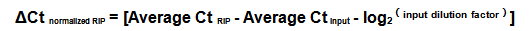
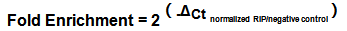
你Get到了m6A-IP-qPCR正确的打开方式了吗?
本文方法参考来自以下几篇文献,若要了解详细的方法,老师可以自行下载对应文献查看。
m6A抗体富集这部分的protocol,请参考m6A权威,m6A-seq的发明人以色列特拉维夫大学医学院的Gideon Rechavi教授发表在Nature Protocols(Pubmed id:23288318)的文章。这里面会有非常详细的步骤教你如何做m6A抗体去IP带有甲基化修饰的RNA。到时候注意打断长度为200-300nt即可,打断需要做几次条件实验。也就是说除了打断长度转换成200-300nt,富集到的RNA从上机测序变为PCR外,其他所有步骤全部一样。文章中的生物信息部分可以忽略不计。
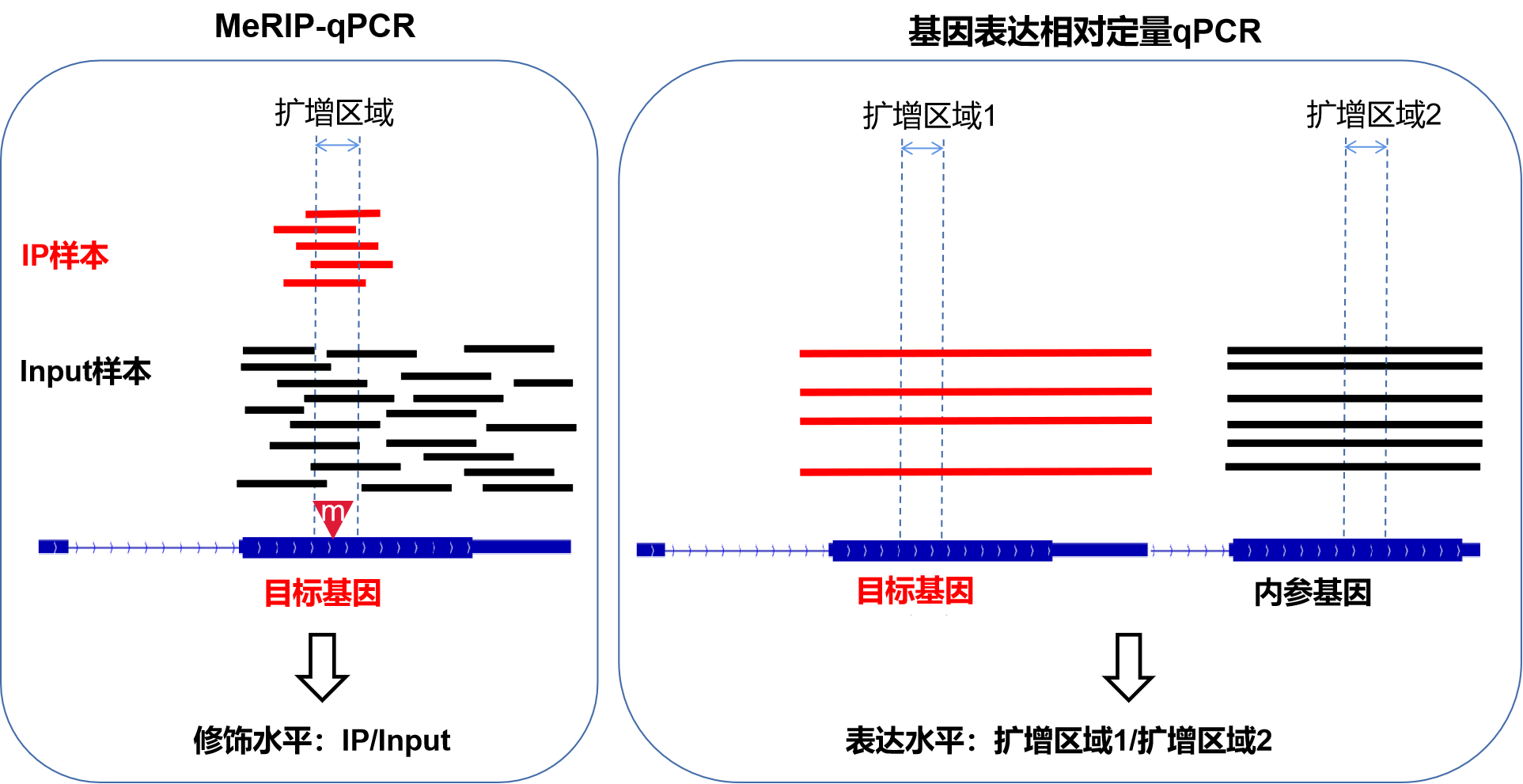
MeRIP-qPCR为什么没有内参基因?
(1)MeRIP-qPCR不需要内参来进行样本间的均一化矫正
从结果的形式%(IP/Input)我们也可以看到,这个实验侧重看修饰的片段(IP样品qPCR结果)占总片段(Input样品qPCR结果)的比例,实验目的只是要得到这两者的比值就体现修饰水平,因此MeRIP-qPCR不涉及到普通的相对定量qPCR需要的内参:
内参是衡量不同样品间表达量时用来均一化样品的,比如默认GAPDH在你的各个样本之间表达量应当相同,qPCR结果样品A中目的基因表达量1,GAPDH表达量10;样品B中目的基因表达量1.5,GAPDH表达量15,那么不能说因为1<1.5所以就说A样品目的基因表达量低,因为从GAPDH来看可能只是实验某环节中A样品的量整体少了一些而已。而是要看A中目的基因表达量是GAPDH的0.1倍,B也是0.1倍,两样品中目的基因表达量其实无差别。
换做MeRIP-qPCR中,如果针对目的基因目的位点qPCR,样品A的IP结果0.2,Input结果1;样品B的IP结果0.3,Input结果1.5,我们的%(IP/Input)结果显示两个样品甲基化水平数值都是0.2,修饰水平无差异,到此就完成了目的。
有人问那没有做内参,万一发生如上段提到的样品用量等环节有差异,会不会影响修饰水平的比较?显然,如果两个样品本来修饰水平相同,那么夸张地想,即使B样品用量由于手抖加了A的10倍,qPCR结果也会IP和Input信号都增长10倍,不影响这个比值即修饰水平。
(2)没有公认在不同样品中修饰水平都会稳定的基因可做参照
前面第1点主要从多个样品比较的角度解释了MeRIP-qPCR为何不需要内参基因。
而有时出现以下情况:1)没有分组对比,只想看下这一个(组)样品中该基因算不算有修饰,此时有个疑问:结果的%(IP/Input)达到多少可算作有修饰?——回答是目前没有一个定值。那么此时难免会希望能有个已知有修饰的参考基因,我们的目的基因如果水平接近或者超过就算有修饰。2)想看看MeRIP实验体系有没有问题,做个已知有修饰的基因当阳性对照看看效果。然而RNA修饰是一个动态的可逆的酶促反应,在同个组织细胞中的水平可能都会随着环境千变万化,不同组织中更是差别很大,与普通qPCR检测基因表达时能找到稳定表达的看家基因不同,MeRIP-qPCR没有一个稳定修饰的基因做参照,目前RNA修饰文章也都不要求有这种参照。
针对上述目的1),可以同时多做一个非特异性IgG抗体的免疫沉淀富集,对比样品本身的IP和IgG是否有显著差异,放在文章中作为该位点有一定程度的修饰的证据。
针对目的2),也可做IgG对照检测体系的特异性。
参考文献:
1. Weng, HY., et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m6A Modification. Cell Stem Cell 22.2 (2017).
2. Ma, CH., et al. RNA m6A methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biology 19.1 (2018): 68.
3. Liu, J., et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nature Cell Biology 20 (2018): 1074
4. Weng, YL., et al. “Epitranscriptomic m6A Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron 97.2 (2018): 313.
5. Gagliardi, M., et al. RIP: RNA Immunoprecipitation. Methods in Molecular Biology 1480 (2016): 73.
6. Dominissini, D, et al. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nature Protocols 8.1 (2013): 176-189.
请问m6a-rip引物是需要针对m6a peak来设计吗?为啥RIP-pcr就是正常的qPCR引物呢?
您好,请问最后MERIP-QPCR结果图需要怎么呈现呢?是IgG和RIP都有吗?怎么作图啊
老师,您好,我想咨询一下,我做了IgG同型对照,的确是有少量扩增。但是有同学做的是没有扩增的。这个扩增原因是什么呀
您好,请问MERIP-qPCR用的引物可以用methprimer里设计的吗?
您好,请问%input的结果是否应该*100呢
您好 不好意思 刚刚看到留言 这个无所谓的,都可以,看你的习惯,标注清楚即可
写得很详尽,非常受用!想请问一下,要是IgG组测不到Ct值,如何校正呢?
这个我也不清楚,按道理IgG组多少会有少量富集的,是足够PCR反应的,是不是您的样本量太少?如果qPCR扩增曲线可以看出有扩增,只是还未到达阈值,可以试试增加循环。