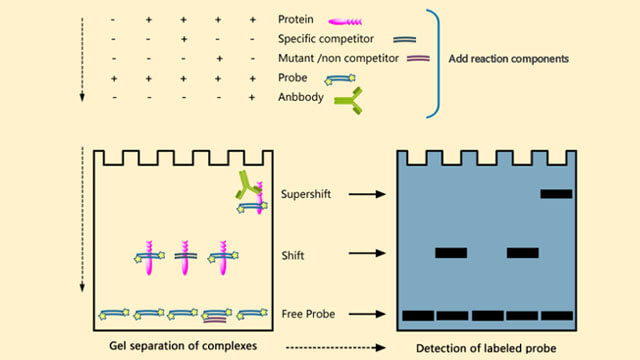
对于实验分组的问题进行简要说明:
问:EMSA实验为何需要设置许多组对照实验?目的何在?
EMSA实验
1,阴性对照反应(标记探针);
2,常规反应(含激活的目的转录因子的核蛋白+标记探针);
3,探针冷竞争反应(含激活的目的转录因子的核蛋白+标记探针+标记探针100倍量的未标记探针);
4,突变探针的冷竞争反应(含激活的目的转录因子的核蛋白+标记探针+标记探针100倍量的未标记突变探针);
5,Super-shift反应(含激活的目的转录因子的核蛋白+标记探针+目的转录因子的特异抗体)。
答:每个对照组的目的:
1.看光有探针,没有其他成分时,探针的位置,应该位于最下方.不然说明探针里面有杂质,影响电泳。
2.这个是看蛋白和探针的结合,是实验目的。
3.看探针结合的特异性.如果冷探针可以竞争结合,阻碍了标记的探针,说明2中的结合是特异的。
4.目的和3相同.突变的冷探针应该对2的结果没有影响。
5.也是判断特异性,这次是看蛋白的特异性,和抗体结合后,就会有super-shift.如果没有,说明是其他的蛋白结合。
目前EMSA实验中常用的探针为非放射性探针,那么探针设计中应该注意的问题有哪些呢?
1.文献查找与分析:
1)如果利用实验者自测的基因序列做EMSA,需要对其进行分析寻找TF结合序列,并与文献进行比对。
2)在文献中可以查到大多数TF的结合序列,但最好进行交叉验证以防出错。
3)需要注意文献发表的探针序列可能并不适合做非放射性EMSA。
2.探针设计:须注意
1)防止探针封闭成环,
2)防止错位配对,
3)排除目标序列以外结合位点的,
4)防止产生空间位阻,
5)适当考虑AT/GC比例,
6)一般EMSA探针只有几十碱基对。探针太长会产生过多结合位点导致结果分析困难,还会产生超级螺旋结构而掩盖结合部位。
3.核酸合成:
1)合成量不能太少,量太少不准确,易产生多余的单链。
2)需要HPLC纯化。
3)如果是RNA探针,要防止降解。
4.生物素或荧光标记:
1)建议采用Biotin或红外荧光标记,尽量不要采用DIG标记。
2)标记只能在探针末端。
3)为提高灵敏度,尽量探针两端都有标记。
5.双链制作:
对配对的单链需进行仔细计算,确保互补链为等当量反应。探针中的多余单链所引起高背景,使非特异带增加,致结果分析困难。
6. 比活性测定:
新制作的探针需要稀释后才能直接用于EMSA。稀释度是通过将新探针做不同稀释度后与已知量探针进行比活性测定来确定的。
7. 制作应用液:
按比活性确定稀释倍数,用TE稀释制作探针应用液,-20保存。
实验中常见的问题:
1、为什么看不到迁移带? 1)蛋白样本提取质量不高,蛋白降解或者提取量不足。 2)样本中没有可以与探针结合的蛋白。 3)探针与蛋白无特异性的相互作用。 4)转膜效率低,蛋白或者探针未转移到膜上。 5)曝光或者成像时间过短。 6) 在Super-Shift EMSA 测定中看不到 Super-Shift DNA/蛋白复合物带
还可能有以下原因:
a. 抗体没有工作。不是所有的抗体都可以用于 Super-Shift EMSA,只有对非变性蛋白的表面抗原决定簇起反应的抗体才能够用于 Super-Shift EMSA。
b. 测定的活化的 DNA/蛋白复合物中没有希望检测的构成成分存在。此时既看不到 Super-Shift 的带,也看不到 DNA/蛋白复合物的量的减少。
c. 使用的抗体过度稀释。一般 10~20 ul 的反应液需要使用 0.5~1 ul 原倍的抗体。
d. 多抗与 DNA/蛋白复合物形成大的聚集物而不进胶。在这种情况下,虽然看不到 Super-Shift 的带,但应当可以看到 DAN/蛋白复合物的电泳带明显减少。
2、为什么实验背景高?
1)曝光或者成像时间过长。
2)封闭时间不足或者效率不高。
3)洗涤效果不佳。
4)实验过程中膜没有一直处于湿润状态。
3、EMSA 测定需要多少量的蛋白与标记的探针?
对每一个特定的结合蛋白和探针,所用的纯化蛋白,部分纯化蛋白,粗制核抽提液需作优化:一般所用纯化蛋白的量在 20~2 000 ug 间,用粗制核抽提液,需要 2~10 ug 蛋白形成特异的复合物。部分纯化蛋白与粗制核抽提液应保存在 -80 ℃、探针应保存在 -20 ℃ 以防止降解。
无论探针或是结合蛋白都应避免多次冻融。
4、用什么凝胶条件将蛋白质/探针复合物和游离的探针分离开?
将结合蛋白或粗制核抽提液和目的探针结合,蛋白/探针复合物和游离探针可在非变性聚丙烯酰胺凝胶中经电泳分离。聚丙烯酰胺的浓度一般为 6%,在特定条件下可用高或低的浓度。也可将 TGE 缓冲液(12.5 mM Tris,pH8.3,95 mM 甘氨酸,0.5 mM EDTA)用于不稳定的蛋白/DNA 复合物。在 4 ℃ 进行结合和电泳实验以阻止不稳定复合物和探针的解离。
5、Poly(dI:dC),非特异性竞争 DNA,特异性竞争 DNA 在 EMSA 测定中的作用?
Poly(dI:dC) 由肌苷和胞嘧啶组成。在 EMSA 反应中加入 poly(dI:dC),可抑制粗制核抽提液中转录调节因子与标记探针的非特异结合。结合溶液中的 poly(dI:dC) 的用量需在正式实验前进行优化,一般用量大约在 0.05 mg/ml 左右。当用纯化的蛋白作凝胶迁移反应时,不必一定加入 poly(dI:dC),如加入,则普通反应中所用终浓度不超过 50~100 ug。对核抽提液,每 2~3 ug 核抽提液用 1 ug poly(dI:dC)。
请问拖尾很严重,看不到结合带和自由探针,蛋白减少2倍后也拖尾,探针减少5倍后虽然不拖尾,但是下面的自由探针也没有了
您好,mutant探针设计有什么较为普遍的原则吗?有些文献全部突变为A,有些文献A-C T-G互换。
进哥你好,EMSA能否用琼脂糖凝胶做,不用设计探针直接用启动子区域和纯化的转录因子蛋白,然后跑电泳,用核酸染料染后显色
我在文献中看到过琼脂糖胶做的EMSA
您好,请问在合成冷探针的时候,冷探针的纯化方式改如何考虑?
您好,请问游离的探针逐渐变淡,但没有结合带咋回事? 蛋白浓度大概1-2uM。
您好,我的EMSA是为了证明DNA和蛋白结合,我没有转膜直接跑完胶染色看的,为什么我最后的迁移条带不出现在同一高度呢?我是DNA量固定不变,随着蛋白浓度上升,到后面条带位置越来越大,现在怀疑是不是DNA能够结合多个蛋白。想咨询一下,还可能有别的什么情况会导致后面的结合条带不再同一位置呢?
我也有类似的问题,但我的探针只有25bp,仅有标记探针的泳道也出现了杂带
王老师您好 我也想请教一下 我们刚开始做这个实验 设置了蛋白的浓度梯度后 发现没加蛋白的组在最下面的条带上方大约一厘米处也还是有一个条带 这个条带每个泳道几乎都有 没加蛋白的组要弱一点但也还是有 然后感觉阳性条带好像都在最上面所以不清楚这个中间的条带是什么 请问您遇到过这种情况吗?可能是什么原因呢?
你好,之前做的时候有结合带出现,后来再做都没有条带了是怎么回事?
您好,我的emsa结果,阴性对照有自由探针,实验组没有自由探针但是也看不到结合条带,冷探针组也没有结合条带,但是有自由探针,这是因为什么,该怎么办呢?
你好,想问一下,我的探针跑出来总是两条条带,位置很近(跑的空探针,没有加入蛋白),是探针里有双链和单链探针吗?怎么避免呢?我用的合成探针的方式和起始DNA量跟我以前的操作是一样的,以前合成标记的探针都是一条,是因为冬季室内温度较低,易形成单链吗?
您好,想问一下在载体构建之前,需要分析以下转录因子的蛋白性质来选择标签,设计截短蛋白吗,那如何分析并判断截断位置那。正在准备做EMSA但是有那些注意事项没有注意到。
您好 意思是要得到不同截短蛋白确定结合位置?建议从uniport上确定蛋白结构域的位置,按照不同结构域进行截短
你好,我转膜后加入化学发光液,用发光仪照一次后,泳道就变黄了,这是什么原因啊
你好,这个我也不清楚 是所有泳道都黄了吗?以前wb有出现过蛋白浓度过高导致曝光后条带变黄的现象
您好,我想请教一下,我用的是纯化后的核蛋白进行所以,用的赛默飞说明书的体系进行反应(加了poly(dI:dC)),我经常做出只要加了冷探针去竞争的用到下面的free probe就会偏弱,相比蛋白和biotin标记的探针有结合的泳道free probe就会偏强,想请教一下这是什么问题引起的?
您好,不好意思 这个情况我也不清楚什么原因
您好,我想请教一个问题:在做EMSA实验的时候要做阴性对照,理论上条带应该是在最下面的,但是我做的阴性对照的时候总是有其他条带的出现,怀疑过是探针纯度不好有其他杂条带(生物素标记),我重新P了探针,用胶回收试剂盒进行的回收,重新做还是出现同样的结果(探针长度350bp),困扰了很长时间,会不会是太长了呢
您好,初步推断,可能是因为这个探针形成了二级结构,这样会出现两条带。解决方法当然是避免二级结构,适当减少长度,只保留motif侧翼100左右,再试试看
你好,我想请教一个问题:用的核提取物做EMSA,那不同分子量大小的转录因子竞争结合目标DNA,胶上会呈现出几条带呢?
请问可以分享一下探针设计的具体做法嘛
您好,我使用的是商品化的探针。关于探针设计,比较简单,通过在线数据库预测蛋白与DNA的结合位点,然后将结合位点左右侧翼总共20-50bp序列合成探针,关于探针具体的要求,参考这篇帖子里的注意事项
非常感谢您的回复,请问有什么推荐的预测蛋白与DNA的结合位点在线数据库嘛?
一般我们研究的是转录因子和基因启动子的结合,这个的话有很多在线数据库可以用,你可以在我网站上搜索转录因子。至于其他的话还有另外一个数据库iDRBP_MMC:DNA结合蛋白和RNA结合蛋白识别数据库,你可以试试看,不行的话咱们再讨论