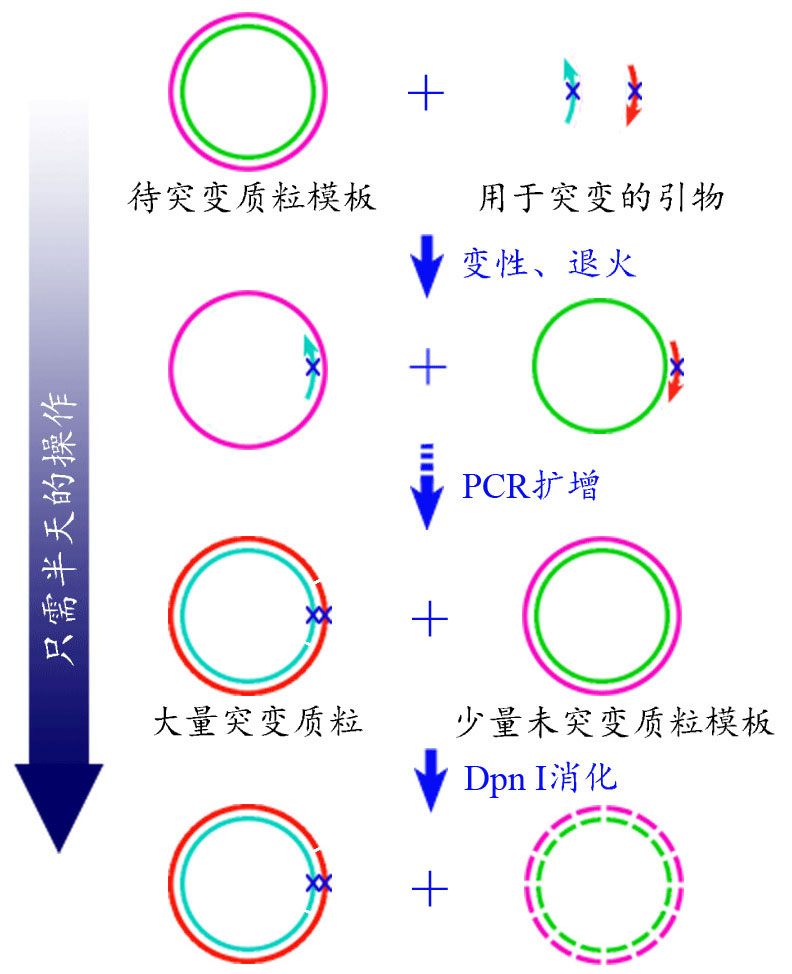
图1.基因定点突变试剂盒原理图
(碧云天:D0206)
使用说明:
1. 引物设计:
用于特定基因突变的引物需要单独设计,请参考如下一些基本原则进行设计:
a. 共需设计两条互补的引物。可以先集中设计一条,然后就可以得到互补的另一条引物。
b. 引物的长度通常为25-45个碱基。
c. 利用如下公式进行引物Tm值的估算,通常Tm值应该不低于78℃。
Tm = 81.5 + 0.41(%GC) – (675/N) – %mismatch
N表示引物所含碱基总数
%GC表示引物中GC碱基数占引物总碱基数的百分值,例如GC含量为50%,那么%GC就是50。
%mismatch表示引物中突变碱基数占引物总碱基数的百分值,例如错配率是2.5%,那么%mismatch就是2.5。
例一:对于模板序列5′-CCAATTTCGAGGAATTAGAACCTTATTTTGAACTTACTGAAGG-3’,其中带有灰色阴影的A为待突变位点,需要突变为C,设计正向突变引物如下:
5′-CCAATTTCGAGGAATTAGCACCTTATTTTGAACTTACTGAAGG-3′
Tm = 81.5 + 0.41*(15/43)*100 – (675/43) – (1/43)*100 = 77.8
例二:对于模板序列5′-GGGAGCTCACCAAGCTGAAGAGCACCTACATTGA-3’,其中两个有灰色阴影的A为待突变位点,均需要突变为G,设计正向突变引物如下:
5′-GGGAGCTCACCAGGCTGAGGAGCACCTACATTGA-3′
Tm = 81.5 + 0.41*(20/34)*100 – (675/34) – (2/34)*100 = 79.9
对于插入、缺失型的突变引物,推荐利用如下公式进行引物Tm值的估算:
Tm=81.5 + 0.41(%GC) ‒ (675/N)
此公式中,N不包含插入或缺失的碱基。因为如果N包含插入或缺失的碱基,数字可能会比较大,从而影响计算结果。
d. 也可以利用如下公式进行引物设计。
引物中突变位点任何一侧都必需满足4*(GC碱基数) + 2*(AT碱基数) ≥ 45。但引物也不宜过长,否则通常会形成非常稳定的二级结构。通常把突变位点两侧的碱基数控制在15个左右,且使两侧按照上述计算得到的数值相近。
例如引物为AGTCAGGCCAATTCGAAGCAGTCGAATTGCCAAG,其中有灰色阴影的AAG为突变位点,则
左侧:4*(GC碱基数) + 2*(AT碱基数) = 4*8 + 2*7 = 46 ≥ 45
右侧:4*(GC碱基数) + 2*(AT碱基数) = 4*8 + 2*8 = 48 ≥ 45
e. 上述c和d这两种方法的计算标准有较明显的差异,但经过多次测试,两种方法都可以顺利获得预期的突变。
f. 在可能的情况下,尽量把引物的GC含量控制在40-60%。
g. 在可能的情况下,尽量使引物不要产生非常稳定的二级结构和引物二聚体。二级结构和二聚体可通过一些软件进行分析。
h. 最好使用经过PAGE纯化的引物或更高纯度的引物。
2. 引物的配制:
如果合成得到的一个引物A的量是20nmol,另外一个互补引物B的量是19nmol。在引物A中加入200μl水,配制成浓度为100μM,在引物B中加入190μl水也配制成浓度为100μM。吸取20μl 100μM引物A和20μl 100μM引物B到一新的离心管中,再加入160μl水,混匀后即可得到可以直接用于基因定点突变反应的引物(10μM each)。
3. 待突变模板质粒的选择:
选择GC含量在40-55%的待突变模板质粒,并且每一个50bp左右的局部GC含量最好也不超过70%。如果GC含量过高,请先把目的基因克隆到其它合适的载体上,再进行基因定点突变反应。另外目的基因GC含量最好也在40-55%的范围,并且每一个50bp左右的局部GC含量最好也不超过70%。如果目的基因GC含量过高,而突变位点不在高GC含量区域,可以先把该基因的不含高GC含量的一个区域克隆出来,进行定点突变,然后再把突变后的片断克隆回原基因中。如果必需使用高GC含量的质粒模板,或者有局部高GC含量的质粒模板,请另外使用专门用于高GC含量模板的PCR反应试剂。
必须使用从dam+的大肠杆菌(这类菌中质粒可以被甲基化)中抽提得到的质粒用于基因定点突变。常用的大部分大肠杆菌都是dam+的,包括DH5α、JM109等。
4. 基因定点突变反应:
a. 如下设置基因定点突变反应体系:
| 扩增片段小于6kb | 扩增片段大于6kb | |||
| 试剂 | 最终浓度 | 体积 | 最终浓度 | 体积 |
| Nuclease-Free Water | – | ?μl | – | ?μl |
| 10X BeyoFusion™ Buffer (with Mg2+) | 1X | 5μl | 1X | 5μl |
| 引物混合物(10μM each) | 0.4μM each | 2μl | 0.8μM each | 4μl |
| dNTP mix (2.5mM each) | 0.25mM each | 5μl | 0.5mM each | 10μl |
| 待突变模板质粒 | 200ng | ?μl | 200ng | ?μl |
| BeyoFusion™ DNA Polymerase | 1/50 | 1μl | 1/50 | ?1μl |
| 总体积 | – | 50μl | – | 50μl |
按照上面的顺序依次加入各种试剂。在上述反应体系中,根据待突变模板质粒的量,计算出需加入的Nuclease-Free Water的量,使总体积为49μl。适当混匀后,加入1μlBeyoFusion™ DNA Polymerase,混匀。如果用的PCR仪没有热盖,在反应体系上加入一滴矿物油(mineral oil)以防止蒸发。
b. 按照如下参数设置PCR仪:
| 步骤 | 循环数 | 温度 | <6kb时的时间 | >6kb时的时间 | 说明 |
| 1 | 1 | 95℃ | 3min | 3min | 最初变性 |
| 2 | 20 | 95℃ | 30sec | 30sec | 变性 |
| 55℃ | 30sec | 30sec | 退火 | ||
| 68℃ | 30sec/kb | 60sec/kb | 延伸 | ||
| 3 | 1 | 68℃ | 10min | 15min | 延伸、补全 |
| 4 | 1 | 4℃ | 长时间保持 | 长时间保持 | 暂时存放 |
说明:上面表格中30sec/kb表示,如果待突变的质粒为6kb,那么68℃的延伸时间为3分钟。60sec/kb的含义相同。
5. DpnI消化:
PCR反应后,直接在PCR反应体系中加入1μl DpnI,混匀后37℃孵育5min。37℃孵育可以在PCR仪上进行,也可以在水浴锅中进行。DpnI消化完毕后可以直接用于转化,或者-20℃保存备用。
6. 转化、挑克隆鉴定:
感受态细菌的转化效率必需至少在107以上,否则很难得到克隆。根据所使用的感受态细菌,加入尽量多的经过DpnI消化后的突变产物用于转化。通常每100μl感受态细菌中可以加入5-10μl经过DpnI消化后的突变产物。按照所使用的感受态细菌的操作方法进行操作,在涂板前通过离心浓缩的办法,把所有被转化后的细菌,全部涂布到含有适当抗生素的平板上,培养过夜。通常会得到100个以下的克隆。如果发现有上千个克隆,那么通常是有什么地方出了问题。
对于得到的克隆,可以挑取3-5个克隆送样测序去测序,以确认得到的克隆是否是预期的突变克隆。通常大部分的是预期的突变克隆。但有时也可能会因为随机因素,会测序了3-5个克隆才得到一个预期的突变克隆。
常见问题:
1. 转化后没有克隆或克隆数极少:
a. 感受态细菌效率不够高,请检测一下感受态细胞的效率,确保转化效率在107以上,当然高一些更好。
b. 把DpnI消化后的产物用常规的乙醇沉淀方法进行沉淀,然后溶解在较小的体积中。这样就可以把所有的产物全部用于转化。
c. 优化基因定点突变中的PCR参数。可以把最初的95℃变性时间延长为5min,循环中95℃变性的时间延长至1min,把循环中的68℃的延伸时间改为1min/kb 至2min/kb,退火可以改为60-55℃或65-55℃等的touch down,退火时间也可以适当延长。
d. 引物设计有问题。通过突变反应中的PCR没有很好地扩增出预期的突变质粒。
e. 如果使用矿物油(mineral oil),在转化时如果把矿物油带入感受态菌,会严重影响转化效率。
2. 有克隆,但没有或很难检测到预期的突变克隆:
a. DpnI消化效果不佳。一种可能是加入DpnI后,由于该酶在甘油中,会迅速下沉,如果没有混匀就会严重影响DpnI的消化作用。另一种可能是DpnI由于保存不当或保存时间过长等原因导致活性下降,这时可以适当延长消化的时间至1-2小时。
b. 使用的待突变的模板质粒量过多,导致DpnI消化时不完全。我们这里推荐的模板质粒用量0.5μg,已经是模板质粒用量的上限,不能再使用更多的模板质粒了。
c. 尽量避免多次反复冻融dNTP。可以把dNTP适当分装后再使用。
3. 有突变克隆,但突变位点不是预期的位点:
a. 引物设计不佳,并且PCR反应中退火温度过低,导致引物退火到错误的地方。
b. 引物质量较差,没有经过PAGE纯化。这样引物中通常会含有比设计的引物要短的特异性较差的引物,容易导致非预期的突变。

您好,做多个点突变的时候,测序结果下来,突变位置正确但是总是紧接着还有一段突变附近区域片段几百碱基的重复,请问这种问题可以怎么改善,PCR各个温度时间都优化进行过延长
这个问题我现在也没法确定,尝试换一下模板DNA,还有减少循环次数,换高保真酶,按照这个顺序分别试一下
转化之后不长菌有没有抗生素的原因,我消化完之后还进行了重组连接,然后转化的适合200ul菌悬液我+了4ulAmp+,在含有氨苄的培养基上,孵育了12小时都没长
你好,我在使用完全反向互补的引物做环状PCR后总会有引物片段的插入,少则1-2次引物插入,多则10次引物插入,我使用的是10 s/kb的快酶,是否是酶的效率太快导致的?
请问为什么我在缺失质粒的时候,pcr扩增时p出来的条带比预期的小很多啊?我预期的大小应该是7000bp,但是p出来只有4500bp左右。Tm值和延伸时间等都考虑了,还是只有4500左右。
您好,我想问一下,我是突变其中的一个氨基酸,设计一对引物后进行PCR扩增整个质粒,扩增之后的产物会自动成环吗,还是说转化到感受态细胞中后它会自动成环,还有就是 是可以直接在PCR产物中加入DpnI的是吗
正向引物和反向引物就是完全互补的关系吗
请问我6000bp的载体做点突,pcr之后跑了电泳,发现亮带是在大于8000bp那里是为什么呢?
DpnI 消化 5min 够吗?我看试剂盒里都是要消化至少 1h
请问,如果想要突变目的序列的最后一个氨基酸,引物设计是否同前
使用什么工具可以评估模板质粒每一个50bp左右的局部GC含量最好也不超过70%?另外,pET-28a(+)适合作为模板质粒吗?
您好,请问DpnI消化后的PCR产物可以直接用来转化吗?是否需要进行纯化
我想检测一下PCR之后是否合成了我想要的点突变质粒,一般电泳上样多少ul,是我拿阴性对照和阳性实验组一起跑电泳,看荧光亮度吗?
请问deletion 的primer 怎么设计啊
你好 请问是个别碱基的delete还是片段?碱基的话就是把突变位置的碱基去掉就可以了,片段的话可以利用同源重组
请问我看有人第二条引物是打乱与第一条引物互补的,这是为什么啊?
你好,没明白什么意思?什么叫打乱与第一条引物互补?
不好意思,这个问题是我就问错了。谢谢
你好,如果想进行多个位点的突变,且位点之间的距离较远,可以同时在一个PCR体系里加入两对引物嘛?如果按照这种方式,是否需要增加循环数?
应该可以的,就是准备多个带突变的引物(同方向,对同一单链模版),延伸时候碰到下一个引物就停止,各片断经连接成环,和单链模版组成杂和环,DpnI消化双链模版,也消化杂和环中的模版,只留下新合成的带多个突变的单链环(mutant ssDNA),得以转化E.coli,形成双链质粒。
最后各个片段的缺口可以经过DNA连接酶连接起来
循环数不需要增加,增加也没有影响
请问是否实验成功
请问点突变之后产物跑胶验证发现整个甬道都有很亮的条带,是什么原因呢?
因为没有突变的被降解了,所以会有弥散的整个泳道都亮,可能就是突变质粒太少或者就是没有突变成功
有构建好质粒,但是选择了PCR分段扩增突变位点。为节省成本,选择不合成,结合目前实验室条件,选择普通PCR做overlap突变,就在突变位点这里测序结果显示未突变。
用常规方式设计引物做PCR插入片段引物突变,随后PCR产物切胶回收,再做overlap PCR连接所有突变片段,但是都没有成功。
嗯嗯 如果构建好质粒的话 不妨用定点突变试剂盒 方便很多;
对于线性的 我当然也会首先考虑设计突变引物分段扩 再进行融合,请问您具体问题是在分段扩增 还是融合这一步
请问为什么我做点突变的时候会出现弥散状的条带,我已经设置退火温度梯度去做了
哪一步的跑胶?
您好,PCR完之后,验证有没有P出来的时候。做了好几次,重新提取质粒、设计引物都试过了
突变PCR这个不需要跑胶验证,如果这个跑胶弥散,我也不清楚,不应该
还是你指的是突变之后 p出突变上下游序列测序验证时发现PCR产物弥散?如果这样,可以拿野生型质粒也P一下 确定是不是引物问题
好的,谢谢!我之后换了一种方法用overlap做点突变了,应该可以做出来。
有构建好质粒,但是选择了PCR分段扩增突变位点。为节省成本,选择不合成,结合目前实验室条件,选择普通PCR做overlap突变,就在突变位点这里测序结果显示未突变。