clusterProfiler是一个功能强大的R包,同时支持GO和KEGG的富集分析,而且可视化功能非常的优秀,本章主要介绍利用这个R包来进行Gene Ontology的富集分析。
进行GO分析时,需要考虑的一个基础因素就是基因的GO注释信息从何处获取。Bioconductor上提供了以下19个物种的Org类型的包,包含了这些物种的GO注释信息
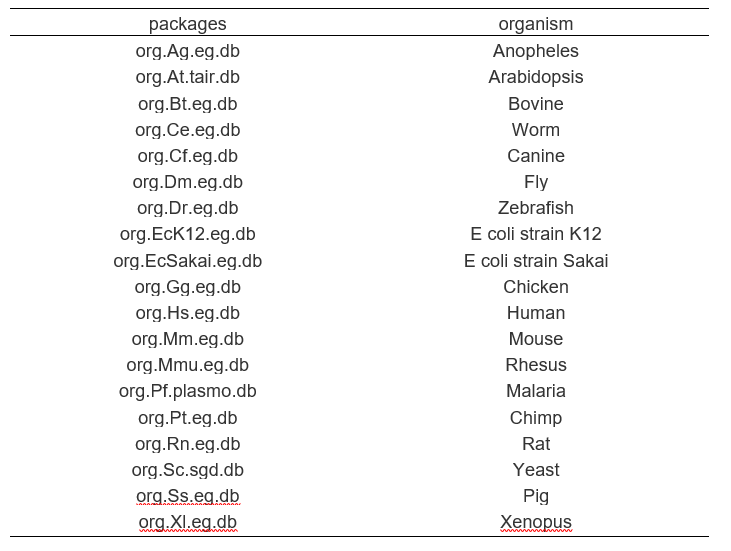
clusterProfiler在做GO分析时,会读取这些包中的GO信息,从这里也可以看出,Bioconductor的生态体系,不同R包的数据传递和依赖组成了其生态环境。
对于以上19个物种,只需要安装对应的org包,clusterProfile就会自动从中获取GO注释信息,我们只需要差异基因的列表就可以了,使用起来非常方便。
在进行富集分析时,支持以下两种算法
1. Over-Representation Analysis
过表达分析其实就是费舍尔精确检验,对于以上19个物种,分析的代码如下
gene就是差异基因对应的向量,keyType指定基因ID的类型,默认为ENTREZID, 该参数的取值可以参考keytypes(org.Hs.eg.db)的结果, 建议采用ENTREZID, OrgDb指定该物种对应的org包的名字,ont代表GO的3大类别,BP, CC, MF; pAdjustMethod指定多重假设检验矫正的方法,cufoff指定对应的阈值,readable=TRUE代表将基因ID转换为gene symbol。
ego <- enrichGO(
gene = gene,
keyType = "ENTREZID",
OrgDb = org.Hs.eg.db,
ont = "CC",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = TRUE)
2. Gene Set Enrichment Analysis
GSEA分析通过置换检验来计算p值,nPerm指定置换次数。
ego <- gseGO(
geneList = geneList,
OrgDb = org.Hs.eg.db,
ont = "CC",
nPerm = 1000,
minGSSize = 100,
maxGSSize = 500,
pvalueCutoff = 0.05,
verbose = FALSE)
对于这19个物种之外的其他物种,也支持读取基因的GO注释文件,然后进行分析,注释文件的格式如下

只需要3列信息即可,第一列为geneID, 第二列为基因对应的GO编号,第三列为GO的描述信息。这3列的顺序是无所谓的, 只要包含这3种信息就可以了。
读取该文件,进行分析的代码如下
对于GO富集分析的结果,clusterProfiler提供了以下几种可视化策略
data <- read.table(
"go_annotation.txt",
header = T,
sep = "\t")
go2gene <- data[, c(2, 1)]
go2name <- data[, c(2, 3)]
# 费舍尔精确检验
x <- enricher(
gene,
TERM2GENE = go2gene,
TERM2NAME = go2name)
# GSEA富集分析
x <- GSEA(
gene,
TERM2GENE = go2gene,
TERM2NAME = go2name)
1. barplot
用散点图展示富集到的GO terms,用法如下
barplot(ego, showCategory = 10) 生成的图片如下
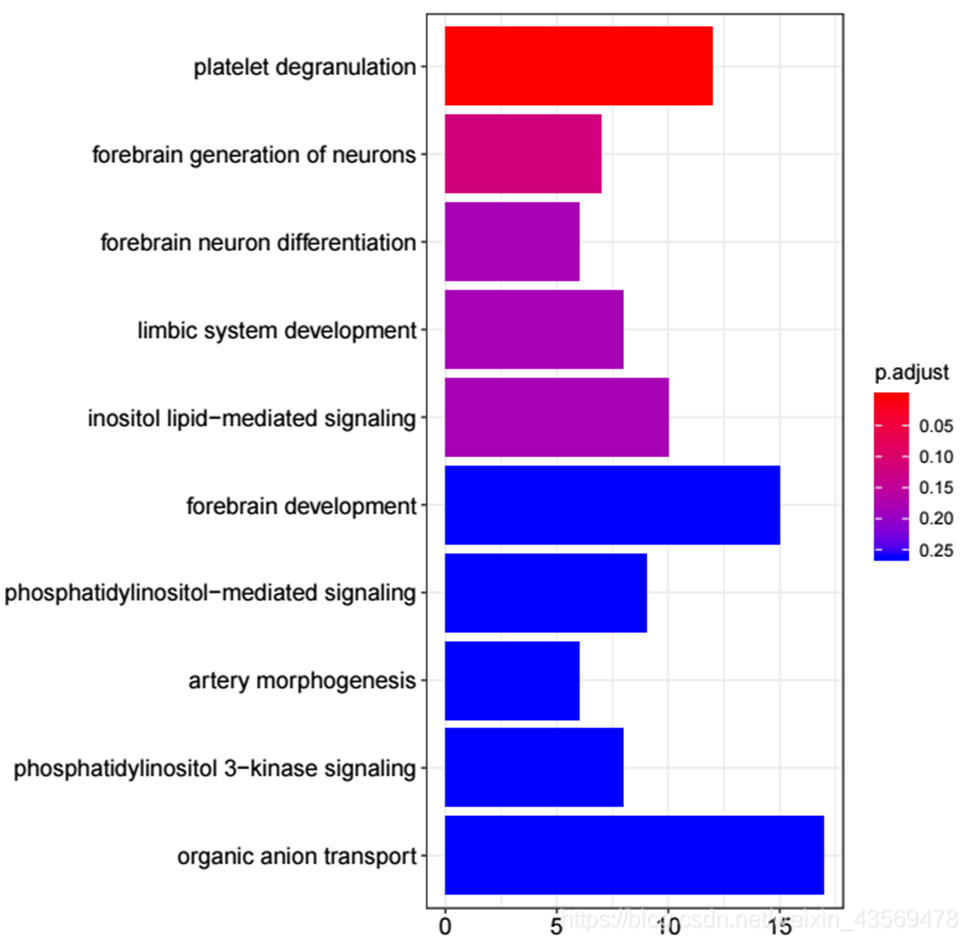
横轴为该GO term下的差异基因个数,纵轴为富集到的GO Terms的描述信息, showCategory指定展示的GO Terms的个数,默认展示显著富集的top10个,即p.adjust最小的10个。注意的颜色对应p.adjust值,从小到大,对应蓝色到红色。
2. dotplot
用散点图展示富集到的GO terms,用法如下
dotplot(ego, showCategory = 10) 生成的图片如下
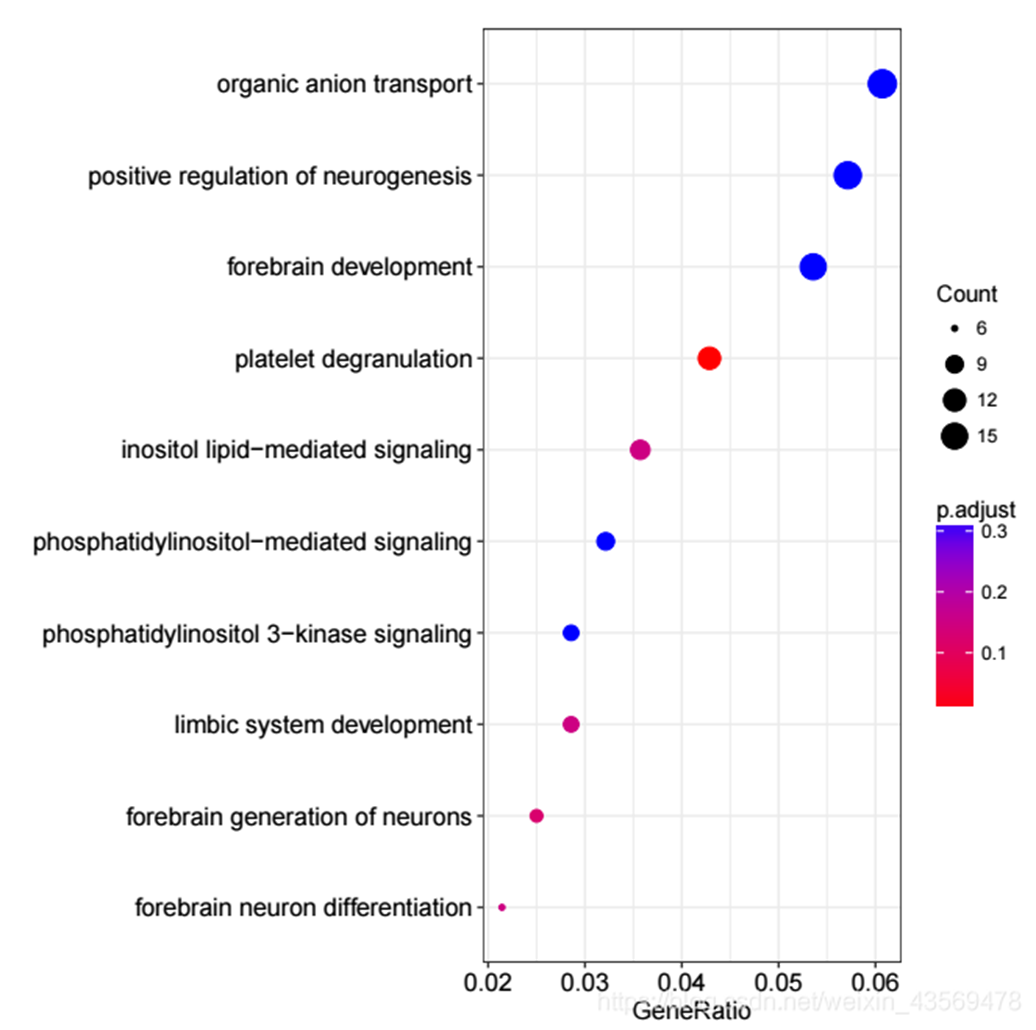
横轴为GeneRatio, 代表该GO term下的差异基因个数占差异基因总数的比例,纵轴为富集到的GO Terms的描述信息, showCategory指定展示的GO Terms的个数,默认展示显著富集的top10个,即p.adjust最小的10个。图中点的颜色对应p.adjust的值,从小到大,对应蓝色到红色,大小对应该GO terms下的差异基因个数,个数越多,点越大。
3. GO有向无环图
调用topGO来实现GO有向无环图的绘制,代码如下
plotGOgraph(ego) 生成的图片如下
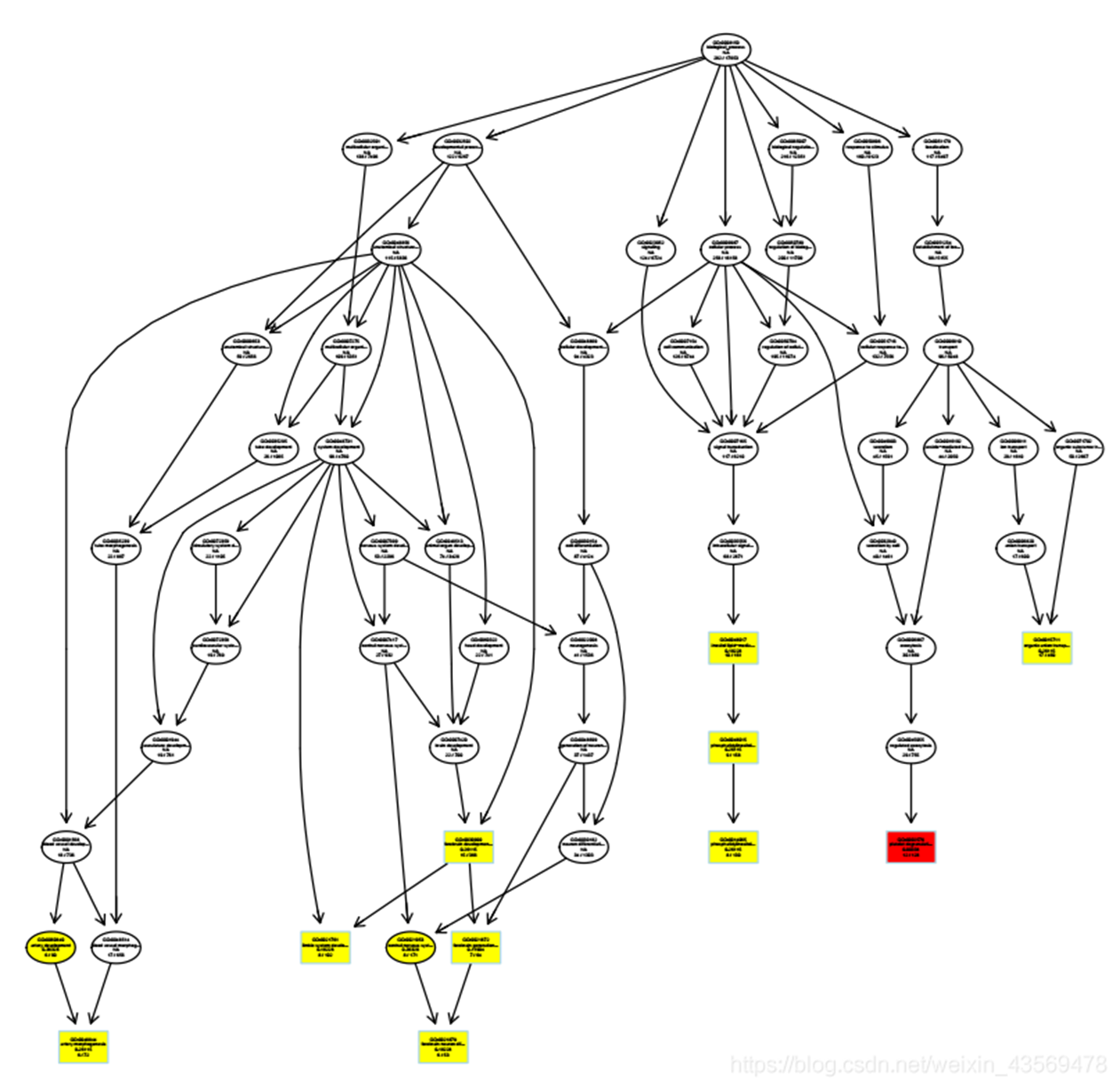
矩形代表富集到的top10个GO terms, 颜色从黄色过滤到红色,对应p值从大到小。
4. goplot
和plotGOgraph展示的信息一样,都是GO有向无环图,只不过采用了igraph中的布局方式,用法如下
goplot(ego)
生成的图片如下

5. emapplot
对于富集到的GO terms之间的基因重叠关系进行展示,如果两个GO terms系的差异基因存在重叠,说明这两个节点存在overlap关系,在图中用线条连接起来,用法如下
emapplot(ego, showCategory = 30) 生成的图片如下
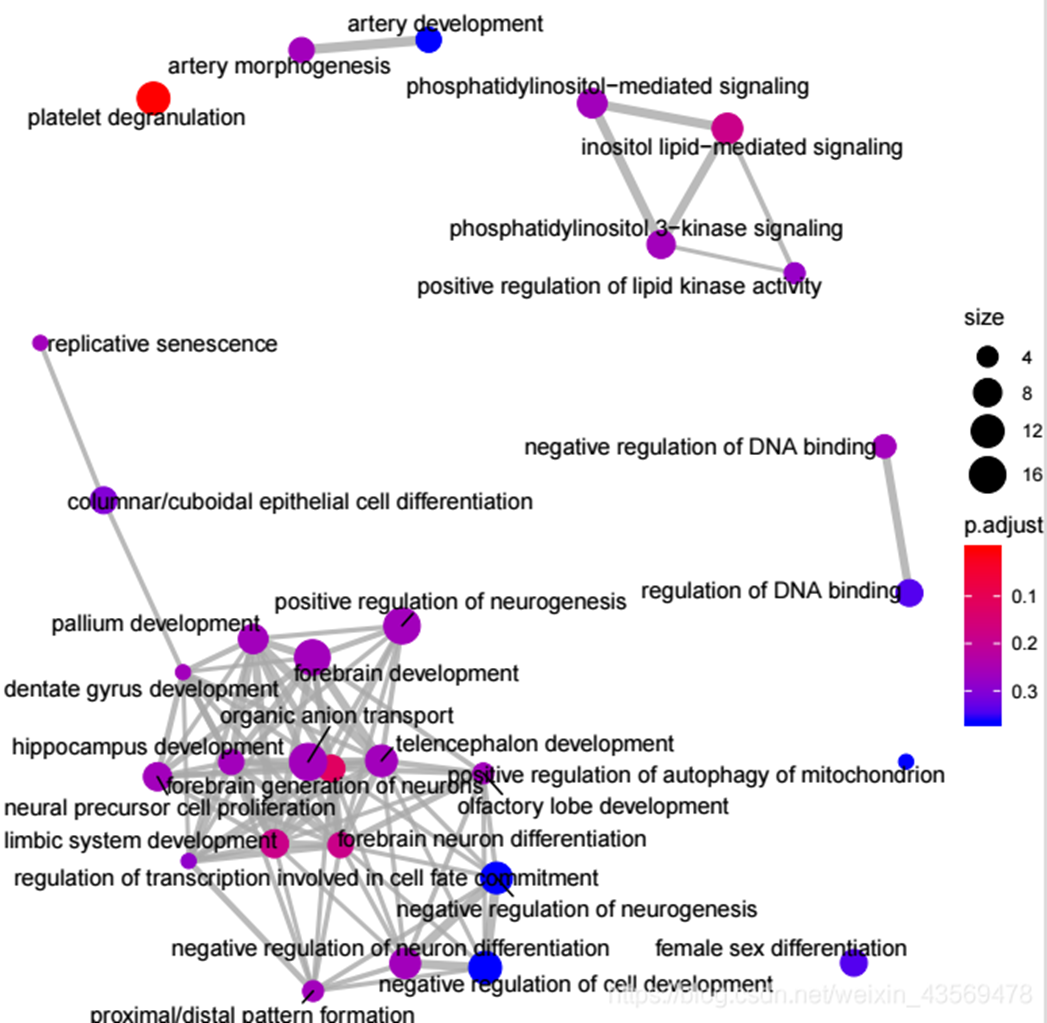
每个节点是一个富集到的GO term, 默认画top30个富集到的GO terms, 节点大小对应该GO terms下富集到的差异基因个数,节点的颜色对应p.adjust的值,从小到大,对应蓝色到红色。
6. cnetplot
对于基因和富集的GO terms之间的对应关系进行展示,如果一个基因位于一个GO Terms下,则将该基因与GO连线,用法如下
cnetplot(ego, showCategory = 5) 生成的图片如下
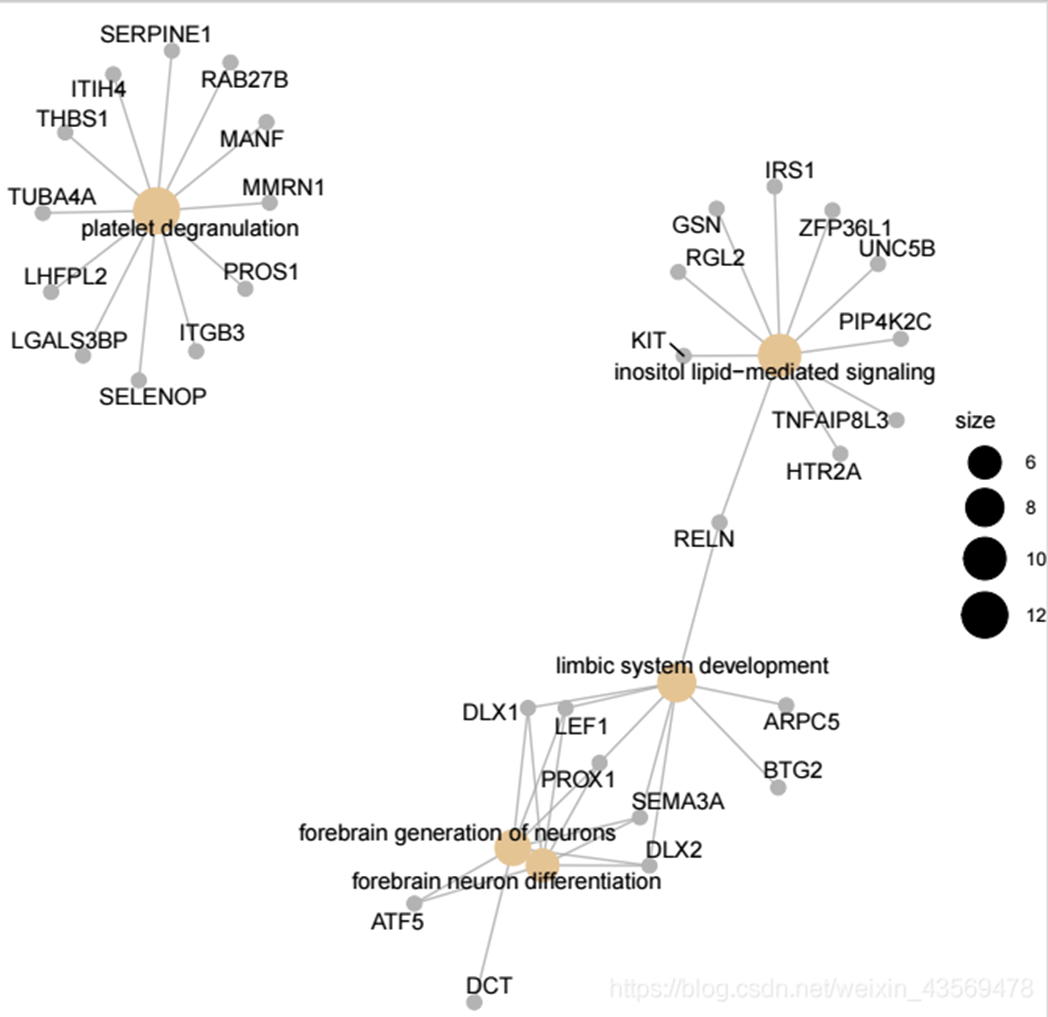
图中灰色的点代表基因,黄色的点代表富集到的GO terms, 默认画top5富集到的GO terms, GO 节点的大小对应富集到的基因个数。更多用法和细节请参考官方文档。
此外进哥最近也在编写基于该包的Shiny应用,有需要的同学敬请期待哦!
这破包依赖太多了