简介
在开发与细胞表面分子结合的基于蛋白质的治疗化合物的过程中,通过受体占用分析(ROA)监测药物与靶点的结合是很重要的。流式细胞术是用来测量大分子治疗性化合物受体占用(RO)的主要技术,因为它可以测量不同细胞亚群的靶向结合。虽然ROA在概念上很简单,但它们是最具挑战性的流式细胞术方法之一。挑战可能与靶标(低水平的抗原表达、稀有群体比例、靶标调节和可溶性靶标的存在)、试剂(用于交叉结合的商业试剂的可用性、内部生产的试剂的开发)以及样品(疾病样本的可用性、样本的稳定性)有关。
在整个药物开发过程中,ROA的应用目的不同。
•在临床前毒理学研究中,ROA被用来确认异种物种中的靶点参与。考虑到异种物种抗药抗体的流行,这些测量尤其重要(Thway等人,2013年)。•当被用作药效学(PD)测量时,ROA数据可以与药代动力学(PK)数据相结合用于建模,以便优化临床试验设计、指导剂量选择,并确定靶向饱和的持续时间和程度(Fisher等人,2016;Quadrini等人,2016;Spilker,Singh,&Vicini,2016;斯特内布林等人,2016;Wyant,Estevam,Yang,&Rosario,2016)。•当与抗药物抗体(ADA)结果结合时,ROA数据用于评估ADA对受体结合的影响,并监测长期受体结合与不良事件的关联。
ROA测量一般有三种类型:结合受体测量、游离受体测量和总受体测量。这些测量也可用于监测对药物结合的反应中的受体调制(Green等人,2016年;Leung等人,2016年;Stewart等人,2016年)。
•结合受体是使用荧光标记的抗药单抗对直接与受体结合的药物的评估。当针对受体的抗体不容易获得时,或者当治疗分子本身的直接荧光素结合可能导致结合特性受损时,使用这种方法。•游离受体是使用竞争性抗体或荧光标记药物来确定未被药物占据的受体。竞争性抗体直接与药物竞争结合同一表位;因此,使用竞争性抗体检测到的任何受体都将不含药物。同样地,这种带有荧光标记的药物只与那些尚未被药物占据的受体结合,可以用来测量游离受体。•总受体测量所有受体,并经常使用针对目标受体的非竞争性抗体。它们将结合到与药物靶向不同的表位,并允许检测所有受体,而不受药物存在与否的影响。或者,可以通过添加过量药物来饱和受体,并使用标记的抗药物抗体来评估饱和的药物结合状态以确定总受体的表达。
受体调节评估治疗药物与其靶受体结合对受体总表达的影响。这可能涉及膜结合受体的上调、下调、内化甚至释放。受体调节评估可以通过监测总受体相对于药物治疗剂量和时间的变化来进行。
使用ROA的类型取决于药物的作用机制(MOA)和试剂的可获得性。如果药物的MOA要阻断与目标受体的结合,游离受体分析通常是首选的形式。当由于试剂的可获得性而不能进行游离受体分析时,另一种选择是结合受体分析,即测量药物占据的受体,然后用来从测得的总受体中推算得出游离受体比例。当药物的MOA涉及受体表达的调节,或药物结合后表达受体的细胞会被去除时,总受体分析是有用的(Liang等人,2016)。
ROA设计和开发
试剂
在大多数情况下,ROA不能仅用商业化试剂来开发。用于检测治疗化合物、抗药抗体(ADA)以及交叉反应和非竞争性的定制试剂可能需要内部生产或由合同实验室生产。在使用定制试剂时,重要的是要确保其管理方式与所有关键试剂一样,并遵循适当的开发、生产和表征指南(King等人,2014年)。在定制试剂投入使用前,应进行特异性检测。当定制试剂与荧光素结合时,试剂的稳定性将是未知的,因此应实施试剂监测计划(CLSI,2020)。所需试剂表征的程度将根据CUU和所需的检测灵敏度而有所不同。最好的做法是建立一种方法来监测试剂降解的迹象,如荧光色素从免疫球蛋白中解离、串联试剂的分解或聚集。
细胞群体选择
药物的MOA通常是决定哪些细胞亚群最适合用于RO评估的主要考虑因素。然而,当有多个合适的靶细胞群体时,ROA应该测量能最有力地测量药物靶标表达水平和靶标参与的细胞群。不同细胞亚群之间的表达水平和其他特征(如靶点的周转)可能有所不同,因此,在给定剂量的治疗分子下的RO测量可能取决于细胞群体(Gavasso,Bringeland,&Tárnok,2019年)。从理论上讲,更高的靶表达水平可能会产生更可靠的检测,但也应该考虑健康捐赠者和患者群体中细胞亚群的频率和稳定性。在某些情况下,在ROA中选择多个细胞群进行评估。例如,Wang等人监测用于PK和PD关联的OX40激动剂ROA时,同时分析了CD4+Tregs和CD4+非Tregs(Wang等人,2019年)
涉及多个靶标
虽然许多治疗分子以单一受体为靶点,但新的药物模式,如双特异性抗体和某些融合蛋白,有不止一个靶点。例如,belatacept(Nulojix®)是一种融合蛋白,将人IgG1的Fc片段与细胞外人类细胞毒性T淋巴细胞相关抗原4(CTLA-4)连接起来。belatacept与抗原提呈细胞(APC)上表达的CD80和CD86均可结合,当开发其ROA时,需考虑几个因素。这种ROA的原因是受体饱和度与剂量频率相关。体外全血实验结果显示,belatacept对CD86的半数最大抑制浓度(IC50)约为CD80的10倍(Latek等人,2009年)。此外,CD80在血液中未受刺激的单核细胞上的表达很低,接近检测下限,而CD86的表达很容易被检测到。因此,CD86被选为ROA靶标。在树突状细胞刺激的混合淋巴细胞反应实验中,CD86饱和被证明是抑制belatacept诱导的同种异体反应所必需的,这一事实支持了这一选择。
可溶性靶标
药物与其膜相关受体的结合可能导致受体的释放或脱落。受体从细胞中脱落会增加可溶性靶分子的水平,进而与治疗分子结合,在形成ROA的过程中呈现复杂的因素。在可溶性受体水平较高的地方,必须评估其对检测抗体可用性的潜在影响。这可以通过在不同水平添加重组可溶性受体(包括预期的最大值)和确定干扰ROA表现的可溶性受体水平来实现。注意,在重组材料的行为与内源性受体的行为有很大不同的情况下,可能需要混合含有已知水平的可溶性受体的血清或血浆样本。
分析前考虑事项
在药物开发研究中,基于流式细胞术的ROA最常见的标本类型是外周全血。与其他流式细胞仪检测一样,抗凝剂、稳定剂和保存温度是检测过程中需要确定的关键参数。在化验开发过程中确定最佳的采样和运输条件将减少验证过程中失败的机会。在一项评估药物随着时间推移与全血中靶细胞结合的研究中,孵育1小时并未导致结合平衡,但在24和48小时后,MFI信号和%ROA可稳定下来。见下图:
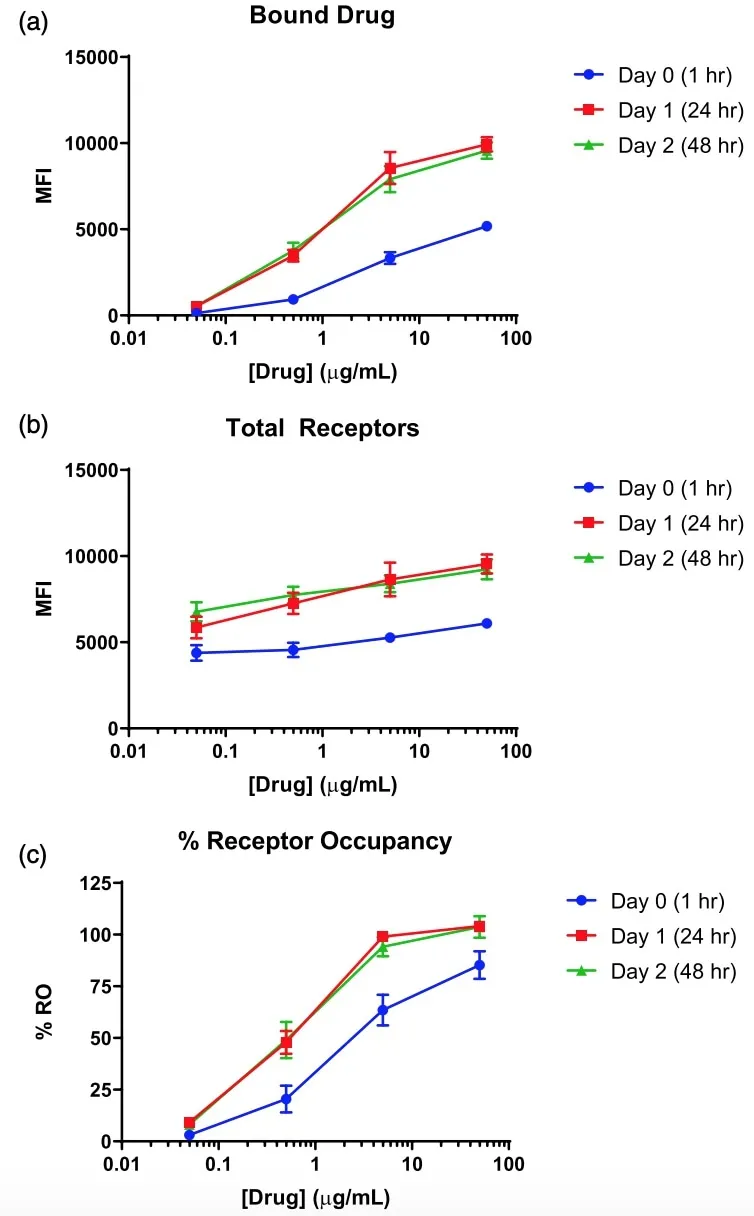
在制定ROA时应同时考虑采血量和运输温度。临床前和临床试验通常需要受试者的大量血液来支持几种分析,包括PK、ADA、生物标记物和RO分析。有一项伦理要求,即最大限度地减少从受试者身上采集大量血液的潜在不良影响,从而将样本采集量降至最低。相反,体积较小的血管(例如,2毫升和6毫升)可能更难使用,导致样本质量较差。样品运输的温度也可能影响ROA的样品完整性(冷包与环境温度),而且大多数血液采集管没有经过冷藏运输条件的验证。
ROA优化
在试剂盒研制完成后,应对其进行优化,以确认该试剂盒是否符合预期。在优化过程中,会评估染色条件,如温度、时间、试剂滴定、细胞浓度和仪器采集参数(Green等人,2016年)。在ROA中,一个关键的考虑因素是评估与测量靶抗原相关的检测的动态范围。这包括在疾病状态样本中检测靶抗原的能力,在疾病状态样本中的表达可能与检测发展过程中使用的健康供体样本的表达不同。最初的实验通常评估至少五种不同药物浓度下的RO,以确定检测的动态范围。在进行ROA时,质量控制(QC)是非常重要的。评估和选择合适的质控材料,以便在验证和随后的样品分析期间进行检测监测,也应在检测优化过程中进行。
样本方面的考虑
当样本可用时,应在优化过程中对患者群体的样本进行评估。通常,ROA的大部分开发工作将使用来自健康捐赠者的样本进行,但重要的是确认该检测将适用于预期使用的患者人群。这可能是一个挑战,因为一些疾病情况很罕见,很难获得。即使可以获得样本,也很难获得新收集的患者样本。当健康状态和疾病状态下的细胞群体和目标表达相同时,就有理由对两组的检测适宜性做出假设。然而,当ROA的靶点在不同的细胞群体上,或者在健康人群和患者人群中表达水平不同时,这些相同的假设可能不成立。
稀有靶细胞群
在某些ROA病例中,靶标表达细胞要么稀少,要么在治疗后变得稀少。例如,当靶细胞耗尽是治疗分子的MOA(例如用于治疗B细胞恶性肿瘤的抗CD20治疗性单抗)时,剂量后样本的靶表达细胞数量可能非常少(Coiffier等人,2008年)。在这些情况下,根据所需的灵敏度将细胞仪设置为获取最小数量的靶事件是一种明智的方法,并确定ROA的量化下限(LLOQ)(Sommer等人,2020年)。分析事件的数量可能不同于检测细胞表型群体所需的事件数量(对于ROA通常较高),并且应该在ROA的开发或优化阶段评估低群体频率对ROA结果的影响以及为目标细胞定义的最小事件数量。
低受体表达水平与靶基因表达调控
分析优化应该考虑受体表达水平非常低的情景,无论是内在的还是由于剂量后下调(Wang等人,2019年)。低受体表达水平可能会降低检测的动态范围和稳健评估RO的能力。在可能的情况下,优化测试应该评估一系列目标表达水平上的RO,以便确定量化的限度。ROA的设计应尽量减少低受体表达对分析性能的影响,因此,用于测量受体表达和衍生RO的试剂应该标记亮的荧光素,并进行适当的滴定。使用荧光素偶联的二抗进行信号放大是改进低水平表达受体检测的另一种选择。方案中的其它设门标记,应尽量选择渗漏少的荧光素,以尽量减少对RO测量至关重要的通道的溢出,避免降低灵敏度。当表位非常接近时,荧光素标记会导致染色抗体之间的空间干扰,甚至产生荧光共振能量转移(FRET)信号(Hlatacek,Posner,&Perelson,1999;Schwarz等人,2019年)。在优化ROA时,可能很难复制在患者样本中观察到的目标受体表达水平的范围;这是一个令人担忧的问题,因为当患者样本的表达水平降至验证期间评估的水平以下时,分析性能可能会有很大差异。这可能反映了药物峰值实验在模拟药物占据的体内行为方面的不足,并可能因受体的实质性调节或抗药物抗体的干扰而变得复杂。在这种情况下,最初的研究数据有助于理解这些关系,并有助于重新开发/重新优化分析,或者在解释数据时创建一种“基于规则”的方法。
受体下调的一个例子是 一种OX40激动剂对CD4+T细胞和Treg的RO评估,该评估是在一项临床试验期间从接受治疗的患者身上收集的样本中进行的(Wang等人,2019年)。该药物被证明在结合后诱导OX40受体内化,令人担忧的是OX40受体内在化会干扰细胞表面受体占有率的测量。验证数据表明,尽管细胞表面OX40的表达水平降低,但仍然可以测量到细胞表面的RO(Wang等人,2019年)。在另一种情况下,受体的调节不是预期的,但明显发生在研究中,并且似乎导致错误的RO结果,可以进行额外的验证测试,以确定该分析不可靠的受体水平。虽然在最初的开发和验证研究中预测这样的情景总是更可取的,但数据仍将为科学论证提供支持,以便从数据分析中消除不可靠的数据。
QC材料评估
确定合适的质量控制材料是优化的关键方面。质控材料可以是保存的全血制剂、冷冻保存或冻干的外周血单核细胞(PBMC),或特征细胞系的冷冻保存等量(Chechowska等人,2019年)。QC材料可以是市面上可买到的,也可以是从供应商处订购的定制材料,也可以是内部开发的。如果是内部开发的,重要的是要确保它们以与所有关键试剂相同的方式进行管理,并遵循适当的开发、生产和表征指南(King等人,2014年)。然后在化验验证过程中验证质控材料的性能。
ROA验证
ROA数据用于内部决策和监管提交。在这两种情况下,都应该进行化验验证,以便充分了解化验的适宜性和性能(CLSI,2020;Green等人,2016)。当像全球临床试验一样在多个地点部署化验时,还应进行化验转移验证(Cabanski等人,2020年)。ROA的验证参数包括线性、精确度和稳定性,并根据需要添加其他评估(Green等人,2016;O‘Hara等人,2011;Piccoli&Michael,2019;Pluim,Ros,Midedea,Bejnen,&Schellens,2019;Selliah等人,2019)。
验证样本
ROA和其他流式细胞仪分析之间的一个主要区别是,必须将不同水平药物添加到基质样本中,以进行分析开发、优化和验证。为此,样品在体外与多个浓度的药物预先孵育,以确保在测试优化过程中确定的反应平衡所需的充足的孵化时间和温度。必须假设替代验证样本模拟体内的情况,在体内药物已经达到与靶点的结合平衡。根据正在评估的验证参数,添加样品的数量和水平会有所不同,但它们应该跨越预期的动态范围,样品处于饱和、中等和低水平的药物结合。
线性度
要验证ROA的线性,需要使用广泛的样本(正常或疾病状态,如果有),这些样本在体外预先孵育,至少有五种药物浓度,从不饱和到目标受体的部分饱和到完全饱和,每个样本都进行了三份测试(CLSI,2020)。这一药物滴定曲线应该包括研究中预期的浓度以下和以上的浓度,并在优化阶段测试的范围内。线性数据将有助于建立分析的动态范围,并可用于补充精密度评估。
精确度
分析内精确度是由一名分析师在单一场合对ROA内的可变性进行的评估。该评估应至少包括三个样本,一式三份。验收标准将由cu确定。对于精确的分析,重复之间计算的变异系数通常可以小于10%,但对于不太丰富的人群,以及异质或模糊表达的抗原,低于25%的变异系数被认为是足够的(Green等人,2016年)。分析间精密度评估分析师在多天和多个仪器上的可变性。这项评估还应包括三个样本,一式三份,在两次或两次以上的分析中进行测试(Fisher等人,2016年)。验收标准将取决于使用环境,然而,在分析场合(仪器、分析员)之间达到低于25%的变异系数的分析可能对大多数使用环境有用(CLSI,2020;O‘Hara等人,2011年;Oldaker等人,2018年;Wood等人,2013年)。较低精确度、较高CV值的化验所产生的数据可能仍对药物开发项目有用,但在这种情况下,应提供ROA是否适合预期目的的理由。
量化下限
当目标细胞数量较低时,必须验证LLOQ并建立报告低于LLOQ的数据的流程(Sommer等人,2020年)。Sommer等人和《流式细胞术进行的CLSI分析验证》(Sommer等人,2020;CLSI,2020)中描述了LLOQ验证的最佳实践。简而言之,验证样本应该用不同级别的可报告结果创建,然后进行一式三份的分析。LLOQ将建立在达到可接受精度的最低值。
QC性能和范围
测定间的精密度也应在质控材料中一起进行。数据可用于确定QC材料的范围(Wood等人,2013年)。
样本稳定性
当样品在采集后不会立即进行检测时,必须对所有化验的样品稳定性进行评估,通常认为在两小时内。应该遵循先前发表的关于流式细胞仪方法稳定性验证的建议(Brown等人,2015;CLSI,2020)。在ROA的情况下,ROA的每个组成部分的稳定性以及最终结果(绑定百分比、自由等),都必须经过验证,包括靶细胞群体的稳定性、靶受体表达水平的稳定性以及结合药物的稳定性都会影响最终样本的稳定窗口。在报告了大量可报告结果的分析中,许多参数的稳定性可能会有所不同。例如,具有双特异性分子的ROA,在不同的细胞类型上,测量两个靶点的RO,每种细胞类型的稳定性以及结合的治疗化合物对不同靶抗原的稳定性可能会有所不同(Vainshtein等人,2016年)。在这些情况下,当一些但不是所有可报告的结果不稳定时,必须在驱动文件中描述处理样本和报告数据的实验室做法。在验证替代验证时,必须在稳定性评估开始时生成添加了治疗化合物的替代验证样本,以便样本最好地模拟临床样本。
实施、传输和监控
基本上,ROA是在新鲜的抗凝全血或骨髓样本上进行的。因此,检测通常在多个实验室进行,该多个实验室是基于在样品稳定性窗口内接收和检测样品的能力而选择的。在这种情况下,除了初始分析方法验证之外,还需要方法转移验证。Cabanski等人描述了方法转移验证的最佳实践。纵向研究中使用的所有分析的最佳做法是建立质量控制系统,以控制和监测过程的所有方面;从样品采集和运输,到处理、仪器校准和数据采集,再到最终的数据分析(Cheechowska等人,2019年)。应遵循分析运行、质量控制验收标准和数据报告的评审和批准的明确流程。文件级别和验收标准由cou和法规要求决定(Oldaker等人,2018年)。关键试剂的批次间的可变性对于客户和许多商业抗体来说都是一个问题。推荐的方法是生产或获得足够数量的关键ROA试剂,以支持整个研究/计划的长度。然而,对于长期的研究或研究范围的改变,这并不总是可行的,继续使用该分析可能需要使用新的试剂批次。当一批新试剂表现出与前一批不同的性能时,需要额外的步骤来统一数据。如果新批次显示更明亮的荧光,可以进行滴定以匹配旧批次的荧光信号,同时保持抗原/抗体平衡。当一批新试剂的荧光信号减弱时,重新偶联可能是最好的方法。或者,在有足够的批次间数据可用或可以产生的情况下,可以实现两个试剂批次之间的数据归一化。
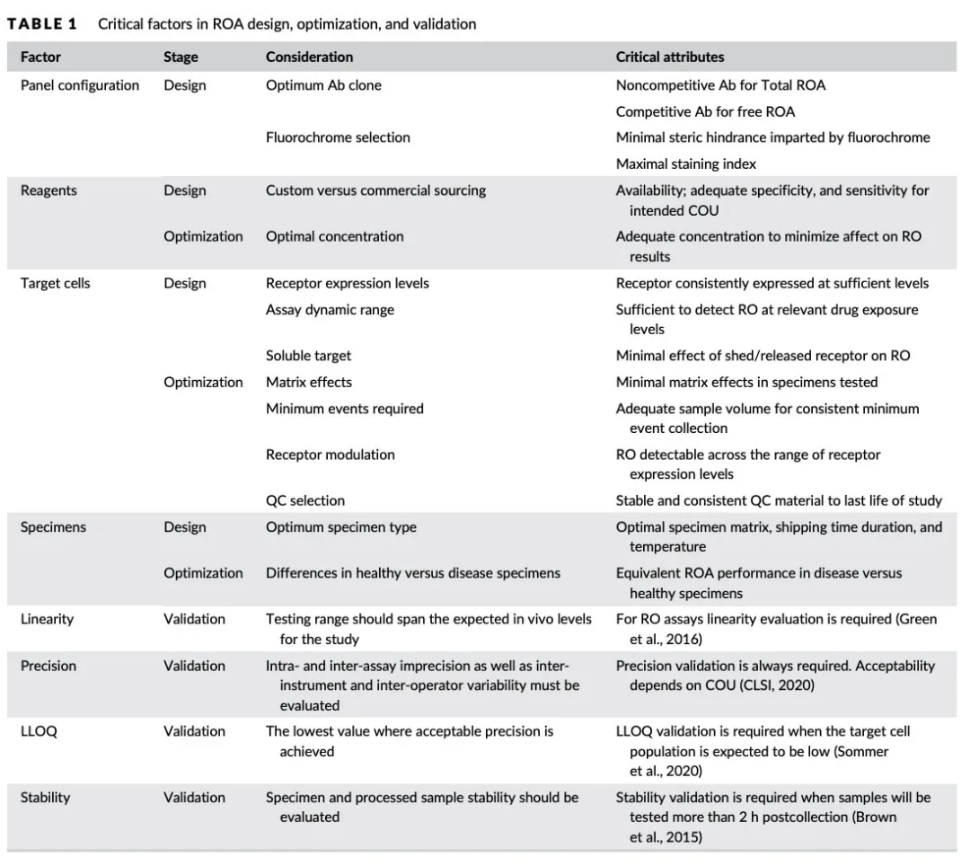
总表
最后,附上文中关键性的这张ROA设计、优化、验证关键因素的汇总表,希望对各位做受体占用率检测的FLOWER有帮助。
翻译自:Hilt E, Sun YS, McCloskey TW, Eck S, McIntosh T, Grugan KD, Lanham DF, Standifer N, Green C, Litwin V, Stewart JJ. Best practices for optimization and validation of flow cytometry-based receptor occupancy assays. Cytometry B Clin Cytom. 2020 Dec 1. doi: 10.1002/cyto.b.21970. Epub ahead of print. PMID: 33259706.