数据库成立背景
由于人口的增长和老龄化,全球癌症的发病率和死亡率正在上升。据估计,2007年全世界诊断出的新病例超过1,200万,并且发生了约760万例癌症死亡事件;如果我们的预防,诊断和治疗癌症的能力没有改善,这些数字将上升到预计的2700万新病例和2050年的1750万癌症死亡人数。癌症对个人,家庭和社会的后果是巨大的。虽然很难估计财务成本,但通过医疗保健系统的直接成本和经济产出损失的间接成本,这些成本也很大。癌症中存在许多病因学因素,包括感染,暴露于化学物质(例如烟草烟雾),饮食,辐射(例如在阳光下)和遗传。虽然这些因素中的一些是可以预防的,但其中很多不是。
介绍
ICGC全称是International Cancer Genome Consortium (国际癌症基因组联合体)。其旨在发起和协调大量的研究项目,其共同目标是全面阐明导致全球人类疾病负担的多种癌症中存在的基因组变化。
ICGC的主要目标是在全球范围内具有临床和社会重要性的50种不同癌症类型和/或亚型的肿瘤中生成全面的基因组异常(体细胞突变,基因异常表达,表观遗传修饰)目录数据,尽可能快地向整个研究团体提供数据,并且以最小的限制,加速研究癌症的成因和控制。 ICGC促进了成员之间的沟通,并为广大科研人员提供了一个平台,达成治疗和预防这些疾病的最大化目标。
ICGC Data Portal提供了用于可视化,查询和下已发布的数据的数据工具。
目前存量
|
Cancer projects |
76 |
|---|---|
|
Cancer primary sites |
21 |
|
Donors with molecular data in DCC |
17,440 |
|
Total Donors |
20,383 |
|
Simple somatic mutations |
68,194,271 |
|
Mutated Genes |
57,668 |
功能操作
进入网页:https://dcc.icgc.org/,可以看到如下图的界面。

然后最直接的就是搜索功能,在搜索框输入你感兴趣的基因,例如BRAF。然后,数据库就会给你一系列与该基因相关的信息。
然后就是,该页面不同的tag。
Cancer Project
点开Cancer Project,所有已经donor的与癌症的相关基因都会如图展示。左边的bar有详细的分别(器官,国家,数据类型,癌症类型)
Advanced Search
高级搜索,基本上就是基本搜索的高级版本,让你可以搜小范围找到你需要的目标基因。你可以详细的选择,该基因的类别(蛋白质,miRNA,rRNA等),Pathway(你可以给出具体的通路),具体的target compounds,Gene Ontology (GO的类别BP,CC,MF),具体的位点(在哪个染色体哪个位置上),等不同的条件。
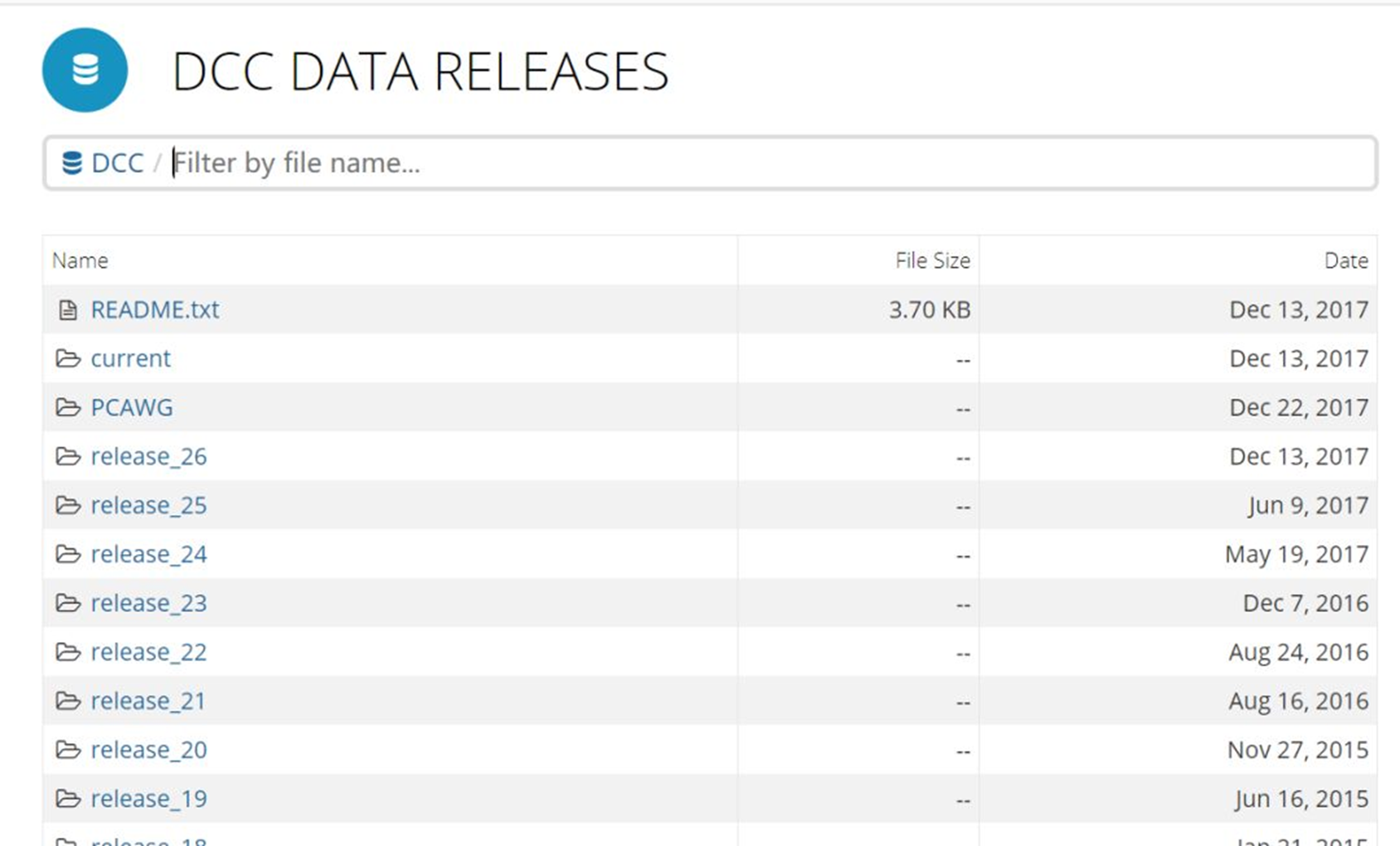
DCC DATA RELEASES
所有目前已经公开的数据如下
Data analysis
数据分析,该数据库也提供一些基本的数据分析,你可以选择一些存储在该数据库的数据,进行一些简单的数据分析。这是这个数据的一个精彩亮点。在数据库里,都有demo示范的功能,可以教大家如何运用这些小工具。
Enrichment Analysis
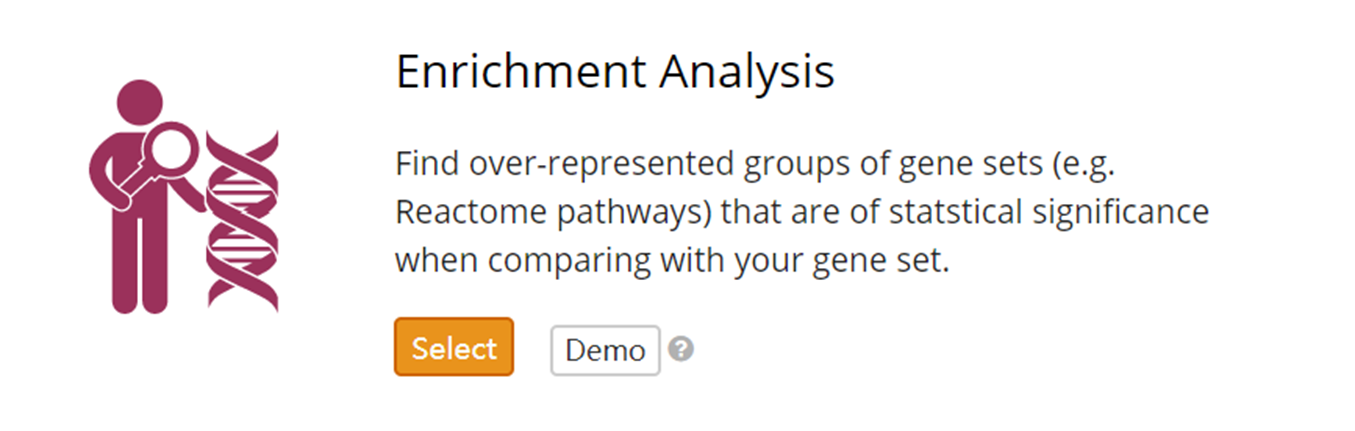
找出与您的基因组比较时具有统计显着性的基因组(例如Reactome通路)的过度代表组。得到相应的GO term 列表。
Set Operations
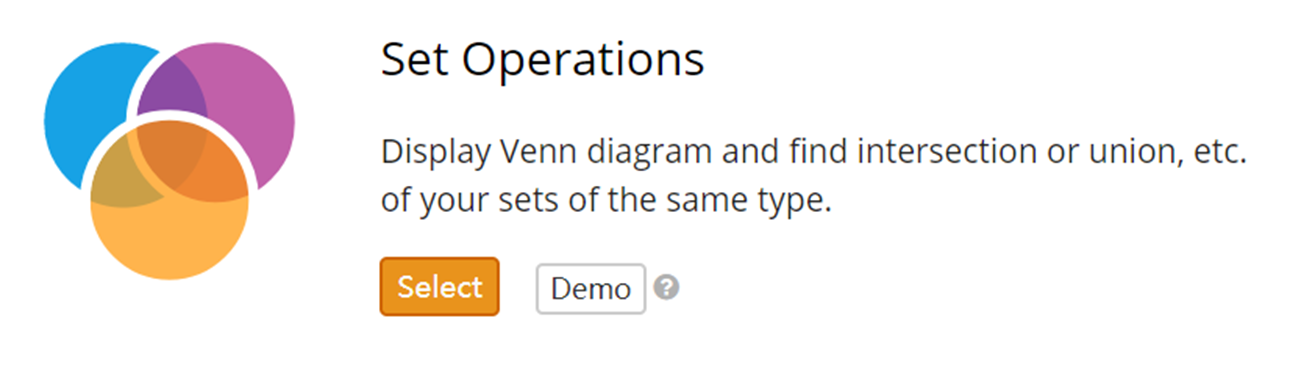
显示维恩图并查找相同类型的基因组的交集或联合等。
Cohort Comparison
显示您的供体组的生存分析,并比较您的供体组之间的性别,生命状态和诊断年龄等特征。
OncoGrid
可视化不同捐助者的基因变化。
数据获取:
下载数据
ICGC下载突变数据
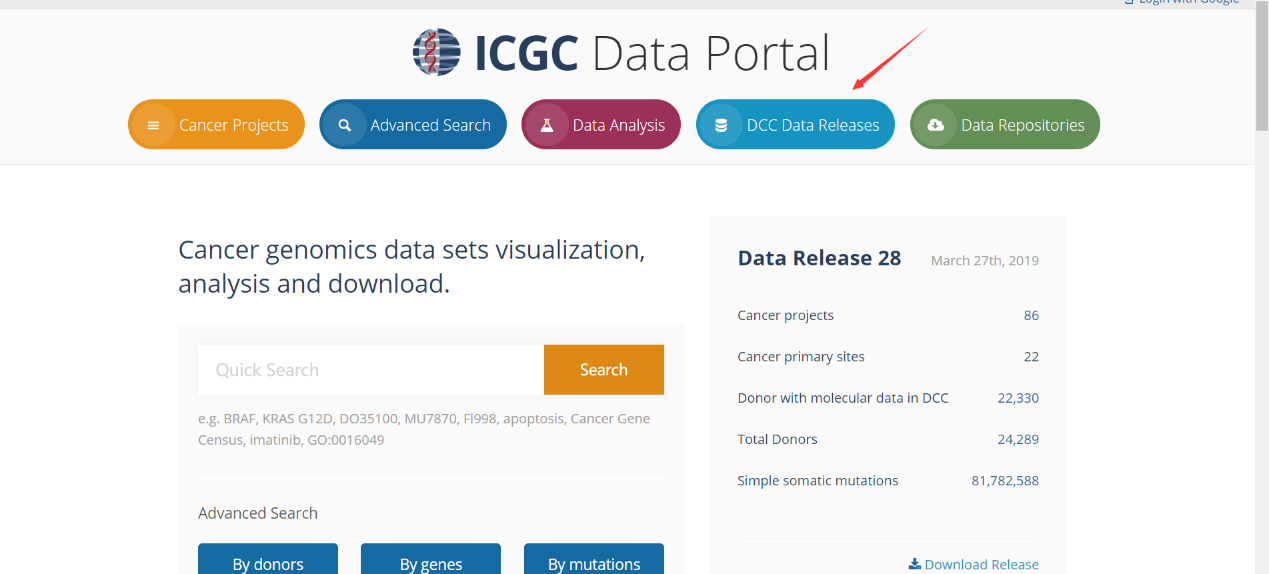
首先点击DCC Data Releases
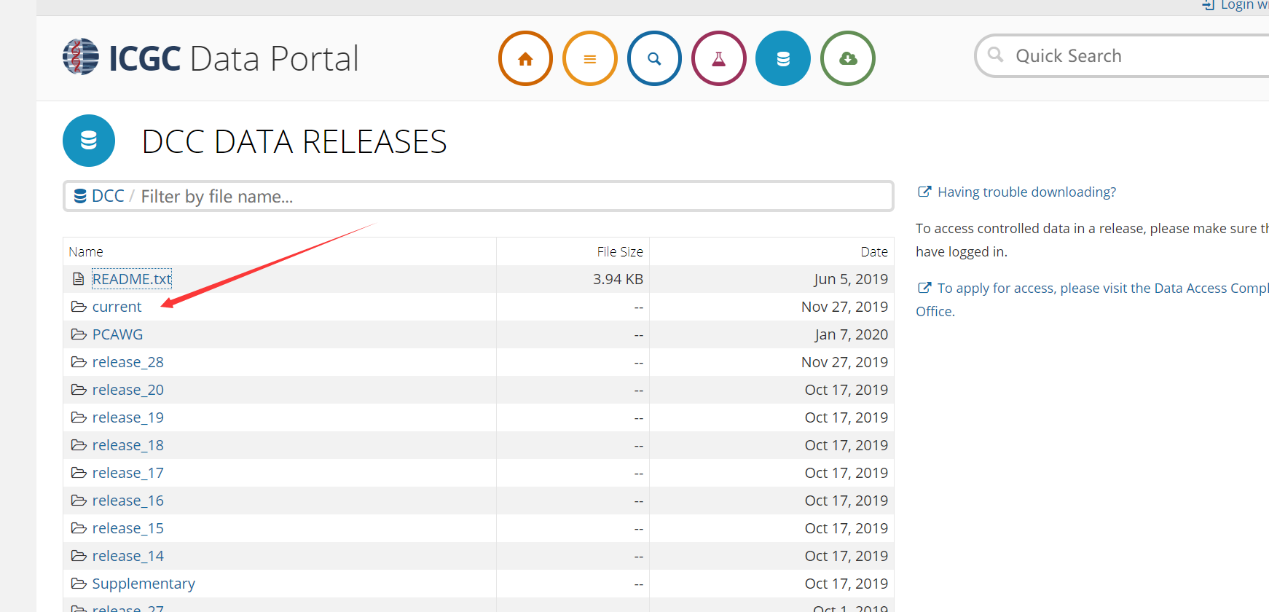
 选择current,下载最新的数据,当然有特殊需求可以下载之前的数据
选择current,下载最新的数据,当然有特殊需求可以下载之前的数据
 然后再点击project就可进入到下图的界面:
然后再点击project就可进入到下图的界面:
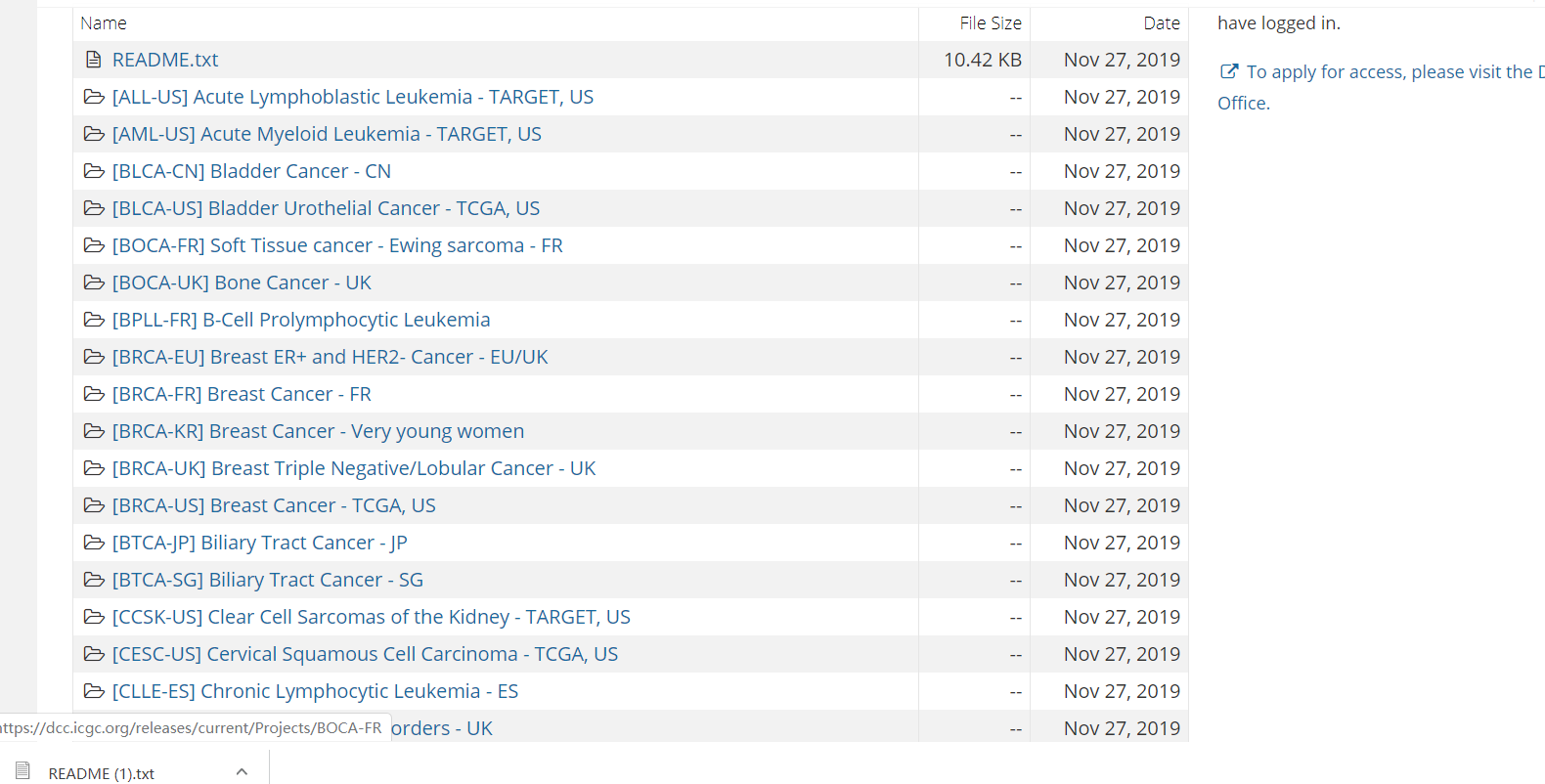 中括号内分别是癌型和地区的信息,大家可以根据自己的研究方向进行选择并点击进去
中括号内分别是癌型和地区的信息,大家可以根据自己的研究方向进行选择并点击进去
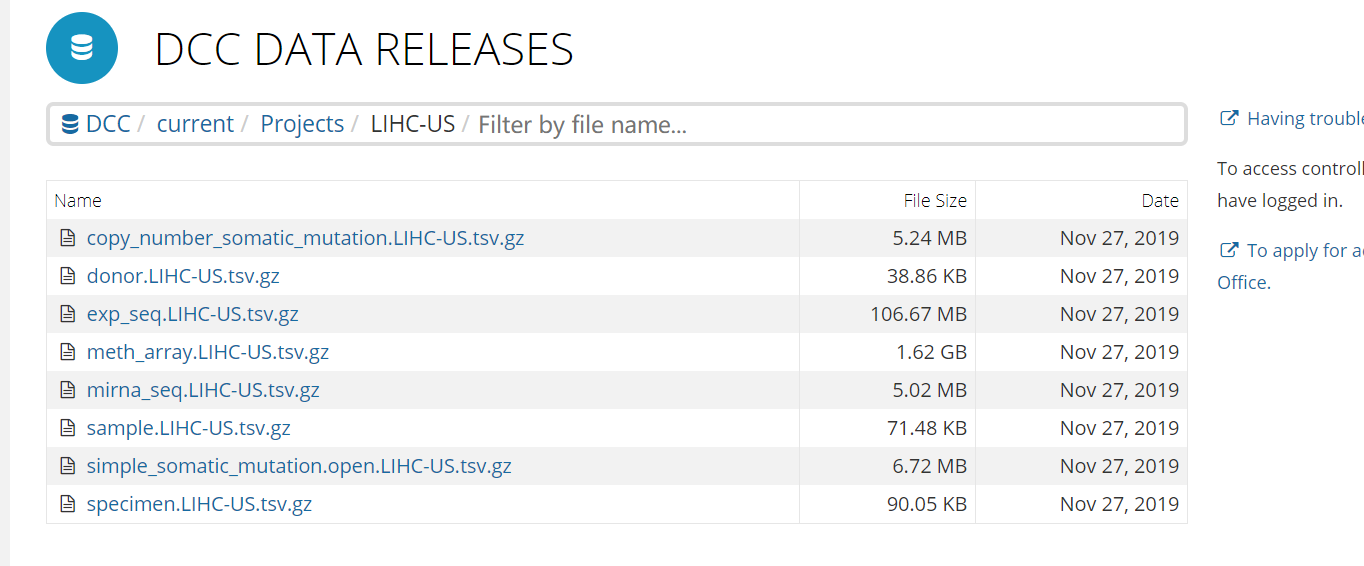 点进去之后可以看到很多不同的数据。不同的癌型不同地区里面的数据类型可能是不一样的。
点进去之后可以看到很多不同的数据。不同的癌型不同地区里面的数据类型可能是不一样的。
一般来说会有拷贝数信息,基因变异信息,临床信息,样本信息等等
选择自己需要的文件点击即可下载,这里我选择了一下三个文件:
 genecode网站下载基因注释文件
genecode网站下载基因注释文件
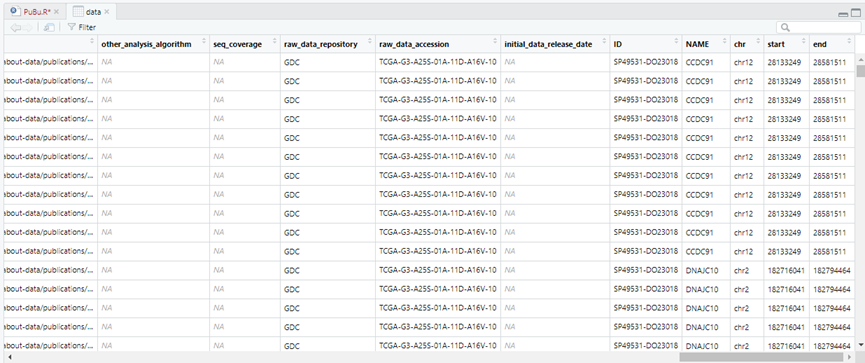
可以使用R包biomaRt完成对基因的注释的(ensembl gene ID –> gene SYMBOL);也可以自己去genecode上下载基因注释文件来完成对基因的注释,并且这个注释文件还有基因的位置信息。
 这样子就下载好了我们需要的基因注释文件啦!
这样子就下载好了我们需要的基因注释文件啦!
数据预处理
读入突变数据
options(stringsAsFactors = F)
data <- read.delim("E:/daiMa/R/puBu/simple_somatic_mutation.open.LIHC-US.tsv", stringsAsFactors=FALSE)
data <- as.data.frame(data)
## 筛选掉不导致氨基酸改变的突变
data <- data[data$consequence_type != "synonymous_variant",]
对data文件进行基因注释
## 制作id,制作突变矩阵的时候要用
Id <- paste(data$icgc_specimen_id,data$icgc_donor_id,sep = "-")
data[,43] <- Id
colnames(data)[43] <- "ID"
## 读入gtf基因注释文件
GeneAnnotationFile <- read.delim("E:/daiMa/R/puBu/gencode.v34.chr_patch_hapl_scaff.annotation.gtf", header=FALSE, comment.char="#")
library(stringr)
## 取出基因注释文件汇总的基因在染色体位置,起始和终止位点
## 这个在画基因突变位点的时候要用
GeneSite <- GeneAnnotationFile[,c(1,4,5)]
colnames(GeneSite) <- c("chr","start","end")
EnsgToName <- str_split(GeneAnnotationFile[,9],";",simplify = T)[,c(1,3)]
colnames(EnsgToName) <- c("ENSG","NAME")
a <- cbind(EnsgToName,GeneSite)
a <- unique.data.frame(a)
## 因为注释文件中基因有许多重复的信息,这里只要带有gene_name的行
a <- a[grep("gene_name",a[,2]),]
a[,1] <- substring(a[,1],9,23)
a[,2] <- str_split(a[,2]," ",simplify =T)[,3]
## 取出突变数据的ENSG编号
ENSG <- data$gene_affected
loc <- match(ENSG,a$ENSG)
na_loc <- which(is.na(loc))
data <- data[-na_loc,]
loc <- na.omit(loc)
data[,44:47] <- a[loc,-1]
write.csv(data,"new_data.csv",quote = F,row.names = F)
注释完成后的data文件: