0.需求
TCGA里的数据tumor多normal少,想要挑选出配对样本进行差异分析。(并不是说必须挑配对样本才能做哦,直接做也是可以的),筛选配对样本解决方案很多种,这边只是一种,本文主要记录配对数据的ggplot2实现。
1.数据
这里使用的数据是本人Xena broower 下载的TCGA-LUAD表达矩阵。
library(ggplot2)
rm(list = ls())
exp<- read.table("G:/TCGA/tcga/LUAD/HiSeqV2",row.names = 1,header = T)
dim(exp)
## [1] 20530 576
exp[1:4,1:4]
##TCGA.69.7978.01 TCGA.62.8399.01 TCGA.78.7539.01 TCGA.50.5931.11
##ARHGEF10L 9.9898 10.4257 9.6264 8.6835
##HIF3A 4.2598 11.6239 9.1362 9.4824
##RNF17 0.4181 0.0000 1.1231 0.8221
##RNF10 10.3657 11.5489 11.6692 11.7341
head(colnames(exp))
## [1] "TCGA.69.7978.01" "TCGA.62.8399.01" "TCGA.78.7539.01" "TCGA.50.5931.11" "TCGA.73.4658.01" "TCGA.44.6775.01"从TCGA ID里可以找到tumor和normal的分组,统计它们的数量:
table(stringr::str_sub(colnames(exp),14,15)<10)
##
## FALSE TRUE
## 59 5172.代码实现
配对样本的共同点是病人ID(前12位)一致,根据这个来匹配即可。
先拆分成tumor和normal两个矩阵,根据ID的14和15位来拆
library(stringr)
exp_nor = exp[,str_sub(colnames(exp),14,15) > 10]
exp_tum = exp[,str_sub(colnames(exp),14,15)<=10]有normal样本的病人的ID,是normal组样本ID的前12位,也是exp_nor的行名。
patient = str_sub(colnames(exp_nor),1,12)然后将tumor矩阵中与patient相匹配的样本名选出来,能匹配到的就会被挑出来。
k = str_sub(colnames(exp_tum),1,12) %in% patient;table(k)
## k
## FALSE TRUE
## 459 58
exp_tum = exp_tum[,k]可以把表达矩阵拼回去咯:
exp2 = cbind(exp_tum,exp_nor)
dim(exp2)
## [1] 20530 117就相当于把表达矩阵按列取子集了,tumor和normal各九个样本,后面可以拿来做配对样本的差异分析
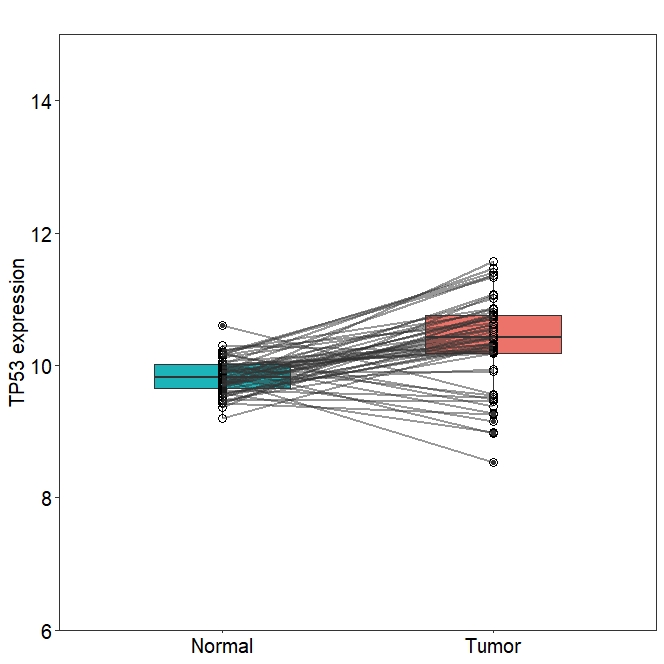
3.特定基因配对T检验即可视化
data<-data.frame( t(exp2[which(rownames(exp2)=="TP53"),]))
data$group<-ifelse(str_sub(rownames(data),14,15) < 10,"Tumor","Normal")
data$patient<-str_sub(rownames(data),1,12)自定义ggplot2主题,可修改参数
if(T){mytheme <- theme(plot.title = element_text(size = 15,color="black",hjust = 0.5),
axis.title = element_text(size = 15,color ="black"),
axis.text = element_text(size = 15,color = "black"),
panel.grid.minor.y = element_blank(),
panel.grid.minor.x = element_blank(),
panel.grid=element_blank(),
legend.position = "none",
legend.text = element_text(size = 15),
legend.title= element_text(size = 15)
)}
box_paired <- ggplot(data, aes(x = group,y = TP53,fill= group)) +
geom_boxplot(aes(fill = group),width= 0.5) +
geom_point(shape=1, size= 3, alpha=1) +
geom_line(aes(group = patient), colour="grey20", linetype="solid",size=1, alpha=0.5)+
scale_fill_manual(values = c("#1CB4B8", "#EB7369"))+
scale_y_continuous(limits=c(6, 15),breaks =seq(6,14,2),expand = c(0, 0) )+
labs(y="TP53 expression",x=NULL,title = "")+
theme_bw()+ mytheme 当然还可以添加paired ttest,使用t.test把结果添加到图中,或者使用ggggpubr包的stat_compare_means函数直接添加。
我输入的data frame是这样的,
id group score
50 tumor 34.6830
51 tumor 55.9853
1 tumor 12.0592
2 tumor 2.5433
没有报错,没有图
当输入plot(“box_paired”) 后报错如下
Error in plot.window(…) : ‘ylim’值不能是无限的
In addition: Warning messages:
1: In xy.coords(x, y, xlabel, ylabel, log) : NAs introduced by coercion
2: In min(x) : no non-missing arguments to min; returning Inf
3: In max(x) : no non-missing arguments to max; returning -Inf
要不加微信吧 网站导航栏个人简历有电话
老师您好,跟您请教下,以上代码运行正常,但是没有出现图形是什么情况呀。是否是样品ID不配对?
您好,这两天忙 刚刚看见 不知道知否解决了,你得对照数据看一下,不清楚可以加微信
我也是没有图出现
图是赋在box_paired 变量中,你得意思是输了box_paired 后没有图?应该不会的,如果有错误会报错的
进哥哥,用配对样本做的结果证据等级是不是稍高一点?
你好,那是必须的 当然前提在于样本量足够,一般TCGA里面配对样本就只有几十个,也足够了,可以配对和两组T检验都做一下