我们经常会在SCI文章里面看到下面这样的图来,展示体细胞突变(somatic mutation)的数据。
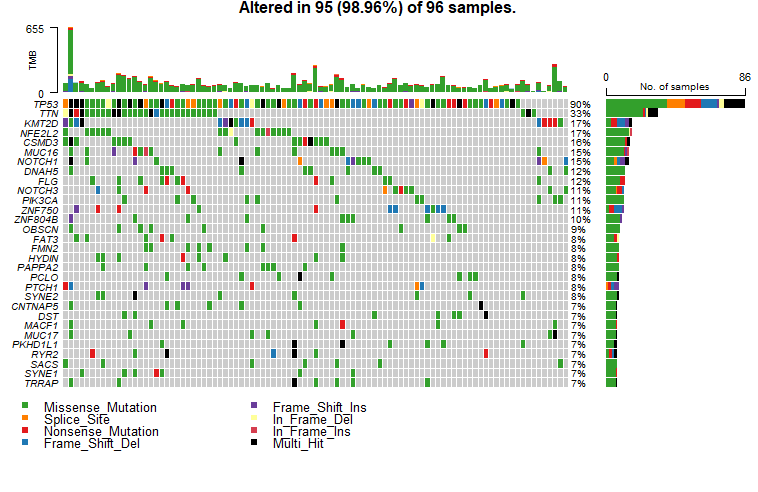
这个图叫瀑布图,展示每一样本中的各种类型的突变,包括错义突变,移码突变,无义突变,插入缺失等等。要想画出这张图,首先我们必须要准本好数据。今天小编就来跟大家聊聊怎么从TCGA数据库下载体细胞突变(somatic mutation)数据。
1.打开TCGA网站,输入需要下载的肿瘤类型
2.点击WXS后面的数字51
3.点击左上角File
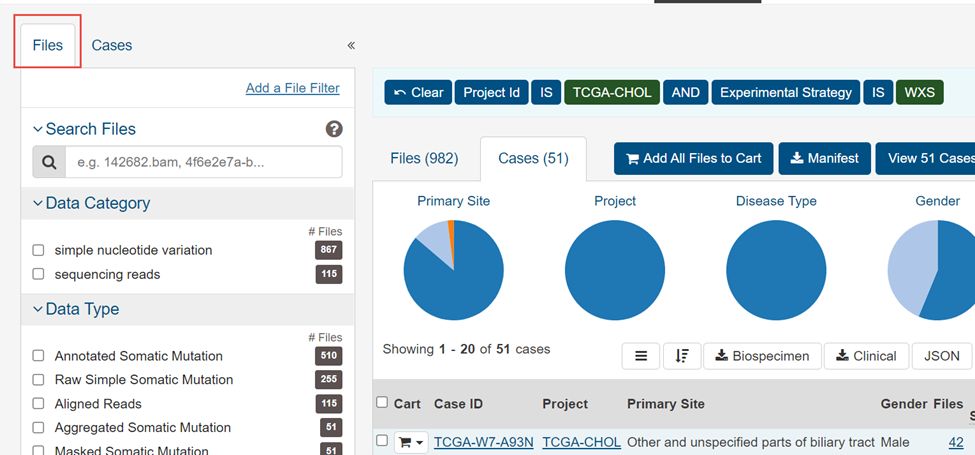
4.选择WXS,Masked Somatic Mutation,maf,simple nucleotide variation,Aliquot Ensemble Somatic Variant Merging and masking,然后Add all files to cart
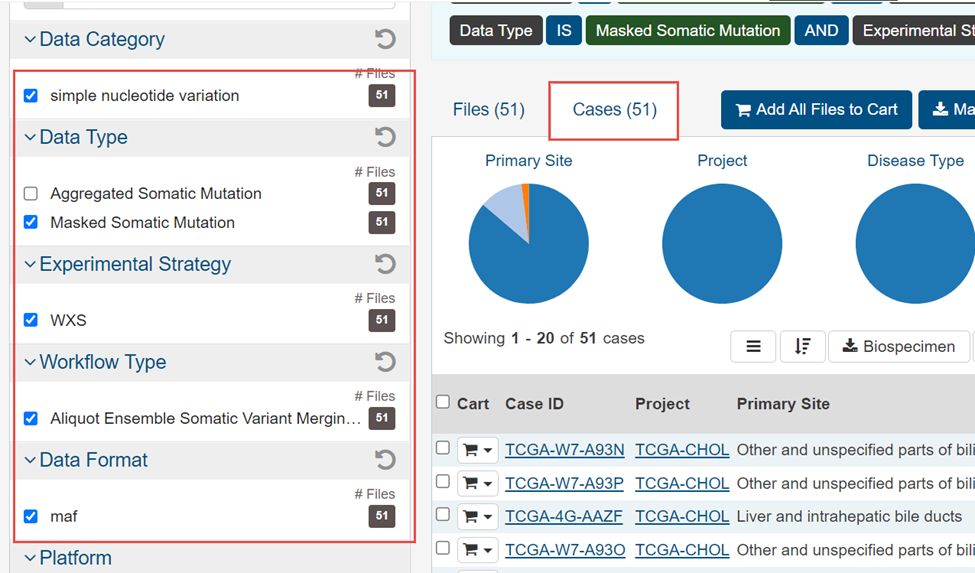
5.这51个文件就加入右上角的购物车里面了
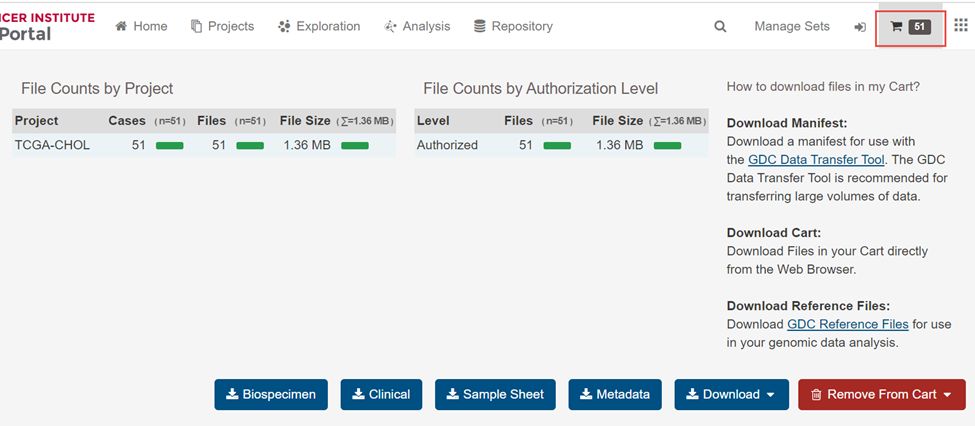
6.下载Download下拉框里里面的Cart
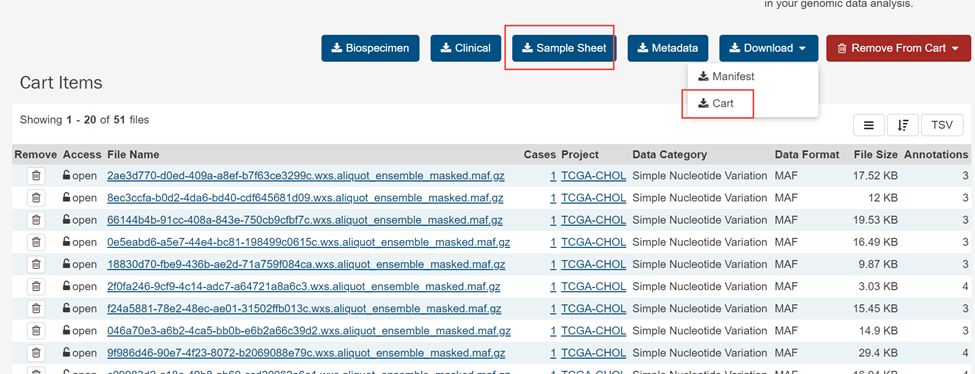
得到gdc_downloa_****.tar.gz.文件
7. 解压该文件
8. 合并所有数据
setwd("G:\\test\\gdc_download_20221025_103238.659115")
files <- list.files(pattern = '*.gz',recursive = TRUE)
all_mut <- data.frame()
for (file in files) {
mut <- read.delim(file,skip = 7, header = T, fill = TRUE,sep = "\t")
all_mut <- rbind(all_mut,mut)
}
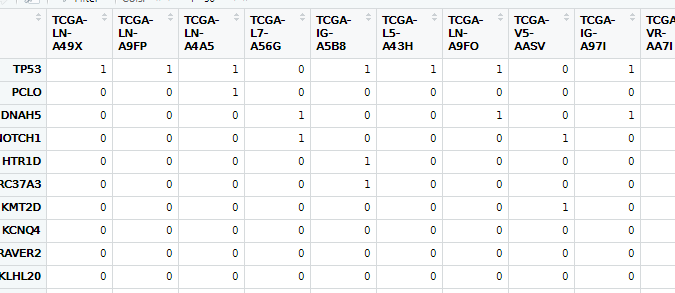
9. 数据整理
all_mut <- read.maf(all_mut)
a <- all_mut@data %>%
.[,c("Hugo_Symbol","Variant_Classification","Tumor_Sample_Barcode")] %>%
as.data.frame() %>%
mutate(Tumor_Sample_Barcode = substring(.$Tumor_Sample_Barcode,1,12))
gene <- as.character(unique(a$Hugo_Symbol))
sample <- as.character(unique(a$Tumor_Sample_Barcode))
mat <- as.data.frame(matrix("",length(gene),length(sample),
dimnames = list(gene,sample)))
mat_0_1 <- as.data.frame(matrix(0,length(gene),length(sample),
dimnames = list(gene,sample)))
for (i in 1:nrow(a)){
mat[as.character(a[i,1]),as.character(a[i,3])] <- as.character(a[i,2])
}
for (i in 1:nrow(a)){
mat_0_1[as.character(a[i,1]),as.character(a[i,3])] <- 1
}
#所有样本突变情况汇总/排序
gene_count <- data.frame(gene=rownames(mat_0_1),
count=as.numeric(apply(mat_0_1,1,sum))) %>%
arrange(desc(count))
gene_top <- gene_count$gene[1:20] # 修改数字,代表TOP多少,也可选择自己感兴趣的
##保存
save(mat,mat_0_1,file = "TMB.rda") ##保存为RData
write.csv(mat,"all_mut_type.csv")
write.csv(mat_0_1,"all_mut_01.csv")

10. 绘制瀑布图oncoplot
oncoplot(maf = all_mut,
top = 30, #显示前30个的突变基因信息
fontSize = 0.6, #设置字体大小
showTumorSampleBarcodes = F) #不显示病人信息
11. 计算tmb值
tmb_table = tmb(maf = all_mut) #默认以log10转化的TMB绘图
tmb_table = tmb(maf = all_mut,logScale = F) #不log
write.csv(tmb_table,"tmb_results.csv")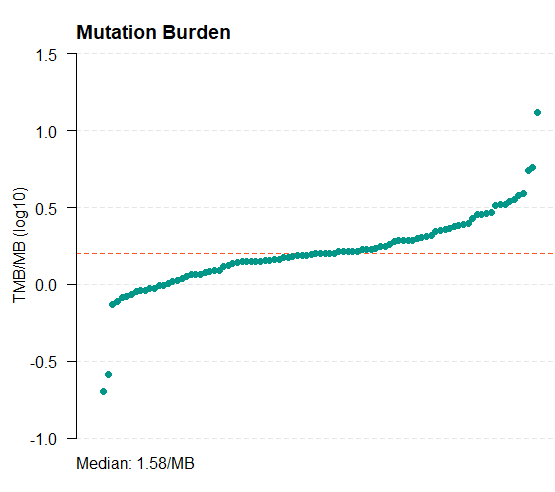
正则表达式匹配那里,可以用加上$,改成*.gz$,表示以.gz结尾,不然有些渠道下载的TCGA的maf数据,解压后会有.maf.gz.parcel格式的文件
是的,我一直懒得更新,有些同学已经遇到这个问题,这里给你置顶,谢谢了
请问第一行截取了TCGA样本名的12位是因为一般处理snp数据均按照单个病人来处理么,如果我是按照基因对样本高低分组来画两组的瀑布图,是不是就可以把12改成16位呀
你好,刚开始接触这部分内容,我有自己测序得到的多个分离物序列,可以做瀑布图吗?
前面整理的哪些矩阵如mat,mat0_1有什么用
您好,请问最后输出的TMB文件,TMB表达量是默认的两位小数吗
进哥哥,你好,我参照你的代码跑了TCGA_CHOL的数据,除了setwd其他都没改,出来的结果和你的不一样,在瀑布图那里我的是Altered in 44 of 51 samples,请问你了解这是哪里出问题了吗?
因為代碼只看Top30,另外沒包含的7個 sample裡面都不在TOP30,你改TOP100就會有了
您好,这个错误怎么解决呀
all_mut <- read.maf(all_mut)
-Validating
Error in validateMaf(maf = maf, isTCGA = isTCGA, rdup = removeDuplicatedVariants, :
missing required fields from MAF: Hugo_Symbol
请问我在整合COAD的WXS时,发现428个病例有457个文件,不是一对一的,这是为什么呢
你好,请问多个maf合并的数据怎样输出到一个maf文件里?
直接rbind即可
老师,能否出一期根据某基因突变与否分组(譬如TP53)行GSEA分析
你好,可以举一反三,在此基础上分析,有问题可以微信讨论
可以吗 谢谢啦!
是分组的瀑布图吗?B站发不了一个小工具介绍 原理很简单
小程序分享TCGA突变maf数据下载整合及分组瀑布图绘制和TMB计算_哔哩哔哩_bilibili
https://www.bilibili.com/video/BV1mN411z7mf/?vd_source=74c93391f1766f275cbe9cbe69ede611
楼主您好,我运行代码的时候运行到arrange(desc(count))
显示Error in arrange(., desc(count)) : could not find function “arrange”,是什么原因?
您好,没有加载dplyr包,这个函数是dplyr包里的。library(dplyr)
咋个在做瀑布图的时候显示特定基因呢
gene<-c("基因list") oncoplot(maf =maf, genes=gene,keepGeneOrder=TRUE) keepGeneOrder=TRUE是否按照你的顺序绘图
进哥,有没有代码例子衔接上下文代码,展示下怎么选定基因进行绘图啊,我用这个提示没有maf
很有帮助,管道符那里之前忘记把
library(dplyr) 声明,问题不大;
有个问题,跑rbind那一段速度太慢了,有无必要提前给数据设定范围,防止R迭代增加导致速度变慢
但是也不好知晓最终合并数据容量有多少(摊手)
您好,如果只是速度慢,可以用do.call,结合future_lapply()并行处理数据,会快很多
设置范围你指的是?没太明白
请问在读取文件的时候报错:
Error in type.convert.default(data[[i]], as.is = as.is[i], dec = dec, :
invalid multibyte string at ‘WS’
In addition: Warning messages:
1: In readLines(file, skip) : line 1 appears to contain an embedded nul
2: In scan(file = file, what = what, sep = sep, quote = quote, dec = dec, :
embedded nul(s) found in input
然后在all_mut <- read.maf(all_mut)这一步报错:
Error in read.maf(all_mut) : could not find function "read.maf"
要怎么解决?
could not find function “read.maf”。。。。
先安装并加载这个包:maftools
BiocManager::install(“maftools”)
library(maftools)
r语言提示不存在 maftools这个包
请问您的第一个问题解决了吗?
若解决的话,请问可以方便给出解析嘛
您好,请问一下保存成mat和mat01两个文件之后,对样本和基因进行筛选完之后怎么重新绘画成瀑布图?
解决了吗 就是用read.maf()读取分好组的数据,后面一样
进哥哥,能出一期微卫星不稳定(MSI)下载分析的教学吗?
好的 尽快整理一下
想请问,TCGA-GBMLGG(GBM和LGG合并)
现在合并后659个样本,给出样本的亚型(659个样本分为高风险组和低风险组)
做这两个组之间的差异瀑布图,可以做到吗按照您的方法?
你好,两组的差异瀑布图?就把两组拆分开分别画就好了,是这个意思吗?
659个样本分为高风险组和低风险组,比如有250个高风险样本,419个低风险样本,分析这两组之间突变基因的差异,并绘制瀑布图,放在一张图里,只不过是高低风险这两组分面展示了,左侧列了差异基因(在高低风险组之间的突变情况是有差异不同的),并且标注了p值
你好 请问你做出分组的图片了吗?
这个需要出教程吗?需要的话我B站搞个
需要
想请教一下绘制瀑布图那里,需要怎么更改代码可以绘制自己的基因
gene_top <- gene_count$gene[1:20] # 修改数字,代表TOP多少,也可选择自己感兴趣的
选择自己感兴趣的基因代码怎么写
这个代码是提取前20个基因名称 如果你有自己感兴趣的 直接gene_top <- c(your genelist)
想请教一下为什么整合完之后样本变少了,415个变成了406个
这是不应该的 如果却是 检查一下代码中的filelist有没有确实 也有可能部分样本没有突变数据