以肺腺癌数据(TCGA-LUAD)为例,为了用TCGA结直肠癌数据做分析,我们首先要先整理出该癌症的基因表达矩阵。(也有一些数据库提供整理好的TCGA癌症数据,如UCSC xena数据库对TCGA数据进行了整理,可直接下载表达矩阵和临床数据用于研究)
进入GDC data portal–>Respository栏目,勾选下面选项:(注意,TCGA更新后的Workflow Type一栏只有STAR – Counts,即将原来的HTSeq-Counts、HTSeq-FPKM、HTSeq-FPKM-UQ数据都放入了一个文件中)
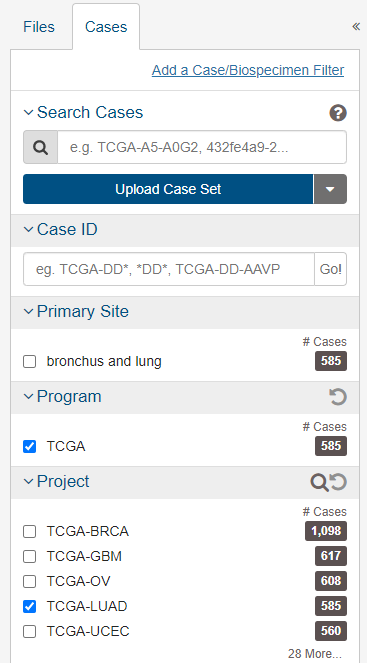
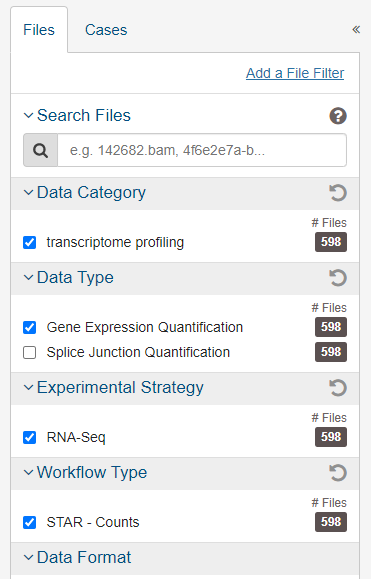
对筛选到的文件,可一键全部添加到cart或手动添加到cart:
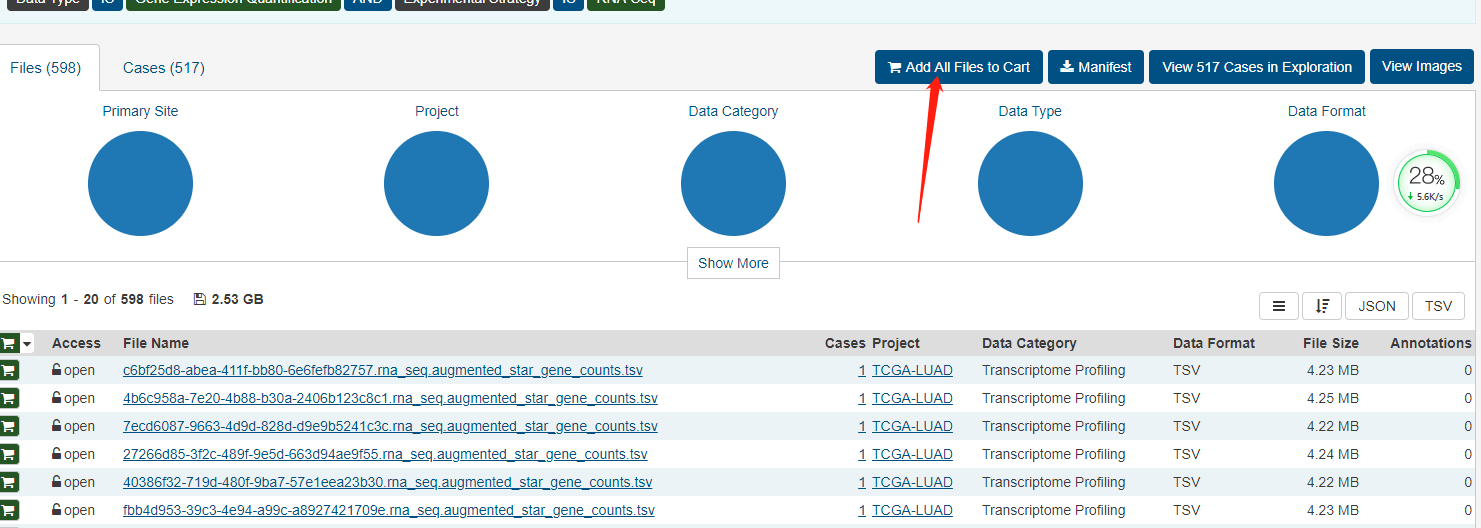
点击顶部Cart,进入下载界面,需要点击这三个地方下载临床数据(Clinical)、json文件(包括文件信息和样本barcode的关系)、表达文件(Download?Cart)。
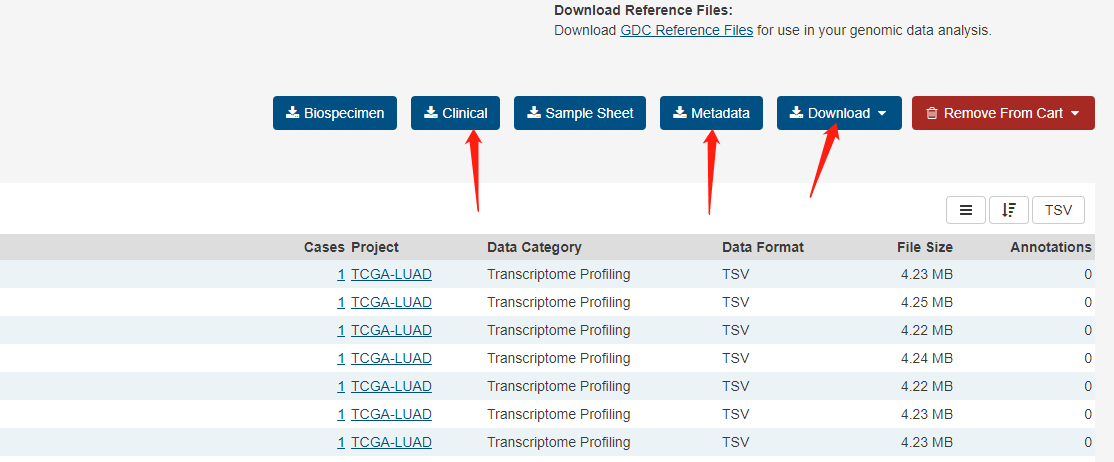
手动解压临床数据文件和json文件,最终我们得到以下三个文件:
到此为止我们下载好了所需数据然后进行数据整理,
Tips: 此处不需要将下载的tsv文件合并到一个文件夹中,如果合并了,会出现样本名称全部为NA
如果已合并,需要对应修改count_file_name <- sapply(count_file_name,function(x){x[2]})为count_file_name <- sapply(count_file_name,function(x){x[1]})
完整代码:
setwd("你的下载数据路径")
#install.packages("rjson")
library("rjson")
json <- jsonlite::fromJSON("metadata.cart.2022-04-18.json")
View(json)
#id <- json$associated_entities[[1]][,1]
sample_id <- sapply(json$associated_entities,function(x){x[,1]})
file_sample <- data.frame(sample_id,file_name=json$file_name)
#获取gdc_download文件夹下的所有TSV表达文件的 路径+文件名
count_file <- list.files('gdc_download_20220418_090958.803273',pattern = '*.tsv',recursive = TRUE)
#在count_file中分割出文件名
count_file_name <- strsplit(count_file,split='/')
count_file_name <- sapply(count_file_name,function(x){x[2]})
matrix = data.frame(matrix(nrow=60660,ncol=0))
for (i in 1:length(count_file)){
path = paste0('gdc_download_20220418_090958.803273//',count_file[i])
data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
colnames(data)<-data[2,]
data <-data[-c(1:6),]
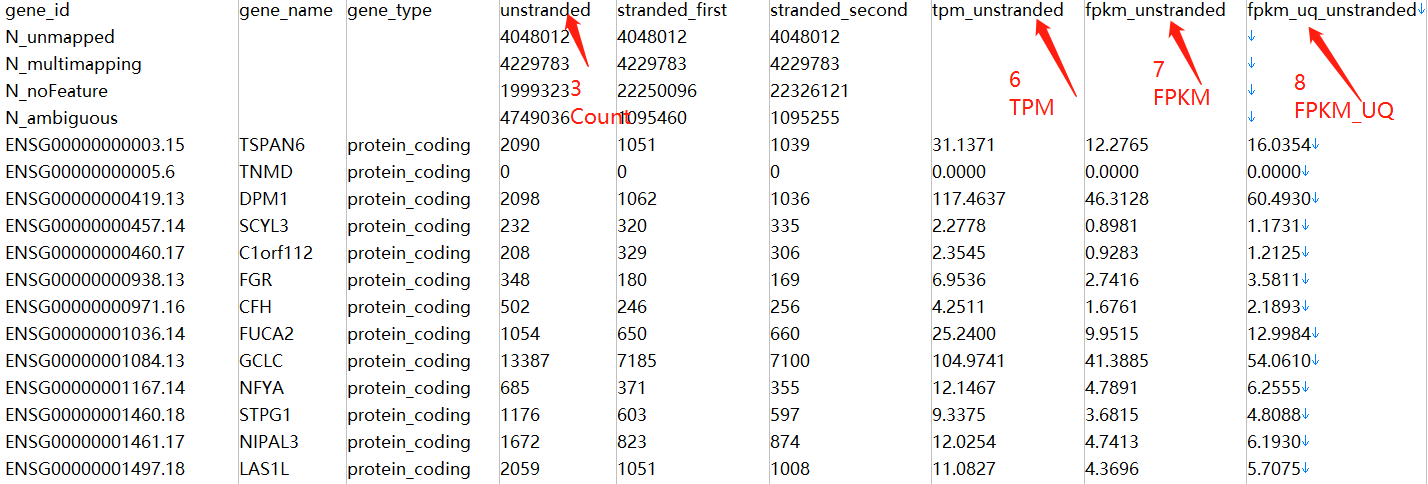
data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
matrix <- cbind(matrix,data)
}
write.csv(matrix,'COUNT_matrix.csv',row.names = TRUE)
设置Gene Symbol为列名
#------------------------------增加部分:设置Gene Symbol为列名的矩阵(前面得到的是Ensembl ID)------------------------------------------
path = paste0('gdc_download_20220418_090958.803273//',count_file[1])
data<- as.matrix(read.delim(path,fill = TRUE,header = FALSE,row.names = 1))
gene_name <-data[-c(1:6),1]
matrix0 <- cbind(gene_name,matrix)
#将gene_name列去除重复的基因,保留每个基因最大表达量结果
matrix0 <- aggregate( . ~ gene_name,data=matrix0, max)
#将gene_name列设为行名
rownames(matrix0) <- matrix0[,1]
matrix0 <- matrix0[,-1]
分为normal和tumor矩阵
#------------------------------增加部分:分为normal和tumor矩阵--------------------------
sample <- colnames(matrix0)
normal <- c()
tumor <- c()
for (i in 1:length(sample)){
if((substring(colnames(matrix0)[i],14,15)>10)){ #14、15位置大于10的为normal样本
normal <- append(normal,sample[i])
} else {
tumor <- append(tumor,sample[i])
}
}
tumor_matrix <- matrix0[,tumor]
normal_matrix <- matrix0[,normal]
#写入文件
临床数据整合
setwd("你的路径")
#install.packages("rjson")
library("rjson")
json <- jsonlite::fromJSON("metadata.cart.2022-04-18.json")
View(json)
entity_submitter_id <- sapply(json$associated_entities,function(x){x[,1]})
case_id <- sapply(json$associated_entities,function(x){x[,3]})
sample_case <- t(rbind(entity_submitter_id,case_id))
clinical <- read.delim('clinical.cart.2022-04-18\\clinical.tsv',header = T)
clinical <- as.data.frame(clinical[duplicated(clinical$case_id),])
clinical_matrix <- merge(sample_case,clinical,by="case_id",all.x=T)
clinical_matrix <- clinical_matrix[,-1]miRNA数据整合
library("rjson")
json <- jsonlite::fromJSON("metadata.cart.2022-09-27.json")
View(json)
#id <- json$associated_entities[[1]][,1]
sample_id <- sapply(json$associated_entities,function(x){x[,1]})
file_sample <- data.frame(sample_id,file_name=json$file_name)
#获取gdc_download文件夹下的所有miRNA表达文件的 路径+文件名
count_file <- list.files('gdc_download_20220927_150057.906231',pattern = '*quantification.txt',recursive = TRUE)
#在count_file中分割出文件名
count_file_name <- strsplit(count_file,split='/')
count_file_name <- sapply(count_file_name,function(x){x[2]})
matrix = data.frame(matrix(nrow=1881,ncol=0))
for (i in 1:length(count_file)){
path = paste0('gdc_download_20220927_150057.906231//',count_file[i])
data<- read.delim(path,fill = TRUE,header = T,row.names = 1)
data <- data[1] #取出count列(第1列),rpm列(第2列)
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
matrix <- cbind(matrix,data)
}
王老师您好,我在运行小程序的时候出现了问题,想请教一下您
问题如下:
Listening on http://127.0.0.1:4140
Error in install.packages : cannot coerce type ‘closure’ to vector of type ‘any’
Warning in body(fun) : argument is not a function
Warning in body(fun) : argument is not a function
Warning in body(fun) : argument is not a function
Warning: Error in registerShinyDebugHook: NULL是不能有属性的
1: runApp
Error in registerShinyDebugHook(params) : NULL是不能有属性的
请问下载gcd压缩文件后,解压一直错误,显示文件损坏,重复下载或者换电脑都不行,这是什么情况啊
为什么将基因名字设置为列名之后矩阵类型变为字符型了,转换为数据型后列名又没有了
normal和tumor矩阵完成之后该怎么做呢
王老师您好,在跑最开始的建立matrix的code 时,出现错误 0052ae83-7ae5-470a-a125-5cd94a9fa9e9/a6a6b9c6-9db7-42b3-a09f-770b7e126fbb.rna_seq.augmented_star_gene_counts.tsv’: No such file or directory 但这个文件在下载的gdc里面时存在的,这个问题如何解决呢?谢谢。
老师,miRNA的也是用这套代码吗,运行到count_file显示character 0,该怎么处理呢
王老师您好, 请问一下 我按照这个代码运行之后 临床数据打开不全 只有几列是怎么回事, 怎样才能获取全部临床信息?
老师您好,
for (i in 1:length(count_file)){
path = paste0(‘gdc_download_20231118_025310.419562//’,count_file[i])时显示:
无法打开文件’gdc_download_20231118_025310.419562//NA’: No such file or directory
朋友这个问题确定了吗,那个60660是确定的嘛?还是需要变动哦
博主您好 运行R包的时候出现了> json <- jsonlite::fromJSON("metadata.cart.2023-11-06.json")
Error in loadNamespace(name) : 不存在叫‘jsonlite’这个名字的程辑包 的错误,该如何解决呢?
请问json包怎么下载呀
> for (i in 1:length(count_file)){
+ path = paste0(‘gdc_download_20231022_140316.972508\\’,count_file[i])
+ data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
+ colnames(data)<-data[2,]
+ data <-data[-c(1:6),]
+ data <- data[3]
+ colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
+ matrix <- cbind(matrix,data)
+ }
😅😅😅Error in file(file, "rt") : cannot open the connection
In addition: Warning message:
In file(file, "rt") :
cannot open file 'gdc_download_20231022_140316.972508\NA': No such file or directory
这个错误不知道怎么办,按照你给的提示改了还是报错,我是下载的THCA 数据
王老师你好,我跑的代码出现了几个问题。问题1 Error in `[.data.frame`(data, 3) : 选择了未定义的列。
问题2 Error in if ((substring(colnames(matrix0)[i], 14, 15) > 10)) { :
参数长度为零
> tumor_matrix normal_matrix <- matrix0[,normal]
Error in matrix0[, normal] : 量度数目不对
另外我还想请教一个问题,我想提取编码蛋白的mRNA去做表达矩阵并进行差异分析,这一步该如何进行?谢谢。
不清楚问题在哪?应该是数据的问题,还没解决的话加微信吧
请问怎么可以加微信啊?
老师,有点不太理解可以加您微信问问吗
进哥,为什么我这个代码出来的character都是(empty)呀
#获取gdc_download文件夹下的所有TSV表达文件的路径+文件名
count_file <- list.files('gdc_download_20231006_123712.253848',pattern = '*.tsv',recursive = TRUE)
解决了吗?没解决加微信
我也是这个问题请问是为什么
同样的问题,最后怎么解决了急
王老师您好,我在运行这句代码是报错了,如何解决,谢谢!
matrix0 <- aggregate( . ~ gene_name,data=matrix0, max)
Error in model.frame.default(formula = cbind() ~ gene_name, data = matrix0) :
参数'cbind()'的种类(NULL)不对
解决了吗?光看着一句看不出来,得看你数据,还没解决加我微信吧
你好,请问这个是什么原因呀,我的路径也没有中文呀
Error: lexical error: invalid char in json text.
clinical.cart.2023-09-17
(right here) ——^
解决了吗 没解决的话加微信看看
王老师您好,我安装rjson包,第5行就报错了
setwd(“D:\\TCGA-LIHC\\transcriptome”)
install.packages(“rjson”, repos = “https://mirrors.ustc.edu.cn/CRAN/”)
#install.packages(“rjson”)
library(“rjson”)
json <- jsonlite::fromJSON("metadata.cart.2023-09-11.json")View(json)
Error in loadNamespace(x) : 不存在叫‘jsonlite’这个名字的程辑包
请您指教
王老师,刚刚那个问题我解决了,安装一个jsonlite包就可以,但是,我又有了新的问题,TCGA下载的数据,解压后不是tsv扩展名,后缀没有counts.tsv,这个肿么办啊
不是RNA测序数据?miRNA的?对应修改后缀即可,还有还需要根据数据结构修改数据列
jsonlite,这包先安装一下
可以问下咋安装吗?去哪里下载?
https://www.jingege.wang/bilibili-code/
我安装了还是报错
进哥,想问两个问题:1、咱们一般做的mRNA是不是就在gene type 里面选择protein coding得到的数据啊;2、是不是所有癌种里面全转录组数据都是60660,然后提取mRNA数据都是19962啊,谢谢!
不好意思,刚刚看见,微信讨论过了是吧
您好,所以一般做的mRNA是不是就是在gene type 里面选择protein coding 得到的数据呢?
另外,提取mRNA数据是19962吗?遇到了和您一样的问题,想打扰您一下,谢谢。
王老师您好,我在运行第四行的时候出现了问题,想请教一下您
问题如下:json <- jsonlite::fromJSON("metadata.cart.2023-08-30.json")#metadata文件名"
Error: lexical error: invalid char in json text.
metadata.cart.2023-08-30.json
(right here) ——^
不好意思,刚刚看见,路径中不能有中文,fromJSON这个函数
路径中没有中文也是这样