这里使用的数据都是已经标准化去批效应的了,具体如下:
各种大型计划产出的RNA-seq数据资源已经非常丰富了,但是大家都想把多个数据库联合起来分析,就不得不面对批次效应这个问题,所以UCSC团队就使用统一的流程把这些数据重新处理了,在亚马逊云上,一个样本花费1.3美元。
发表在:Nature Biotechnology publication: https://doi.org/10.1038/nbt.3772IF: 68.164 Q1
3大数据库是:
- The Cancer Genome Atlas (TCGA)
- Genotype-Tissue Expression (GTEx)
- Therapeutically Applicable Research To Generate Effective Treatments (TARGET)
而且还提供网页工具供查询使用:
Differential gene and isoform expression of FOXM1 transcription factor in TCGA vs. GTEx
使用的数据处理流程
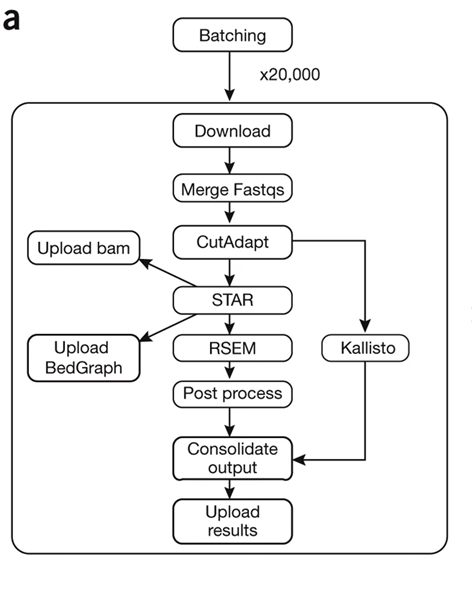
如下图: CutAdapt was used for adapter trimming, STAR was used for alignment, and RSEM and Kallisto were used as quantifiers.
参考基因组选择
- STAR, RSEM, and Kallisto indexes were all built with the same reference genome. HG38 (no alt analysis) with overlapping genes from the PAR locus removed (chrY:10,000-2,781,479 and chrY:56,887,902-57,217,415).
- ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_genbank/Eukaryotes/vertebrates_mammals/Homo_sapiens/GRCh38/seqs_for_alignment_pipelines
- ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_genbank/Eukaryotes/vertebrates_mammals/Homo_sapiens/GRCh38/seqs_for_alignment_pipelines
注释文件的选择
- RSEM: Gencode V23 comprehensive annotation (CHR)
- http://www.gencodegenes.org/releases/23.html first row
- Kallisto: Gencode V23 comprehensive annotation (ALL)
- http://www.gencodegenes.org/releases/23.html second row
- http://www.gencodegenes.org/releases/23.html second row
软件参数的选择
- STAR
- sudo docker run -v $(pwd):/data quay.io/ucsc_cgl/star –runThreadN 32 –runMode genomeGenerate –genomeDir /data/genomeDir –genomeFastaFiles hg38.fa –sjdbGTFfile gencode.v23.annotation.gtf
- Kallisto
- sudo docker run -v $(pwd):/data quay.io/ucsc_cgl/kallisto index -i hg38.gencodeV23.transcripts.idx transcriptome_hg38_gencodev23.fasta
- Kallisto index that was used during the recompute is available here.
- RSEM
- sudo docker run -v $(pwd):/data –entrypoint=rsem-prepare-reference jvivian/rsem -p 4 –gtf gencode.v23.annotation.gtf hg38.fa hg38
- sudo docker run -v $(pwd):/data –entrypoint=rsem-prepare-reference jvivian/rsem -p 4 –gtf gencode.v23.annotation.gtf hg38.fa hg38
Raw data
Nature Publication Supplementary Note 7 – Data Availability
Submitter sample ID to Xena sample ID mapping
最后公布的可供下载的数据集
- GTEX (11 datasets)
- TARGET Pan-Cancer (PANCAN)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (12 datasets)
- TCGA and TARGET Pan-Cancer (PANCAN)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (4 datasets)
- TCGA Pan-Cancer (PANCAN)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) (10 datasets)
- TCGA TARGET GTEx (13 datasets)
其中TCGA TARGET GTEX 3大数据库) (共有 13 DATASETS)
cohort: TCGA TARGET GTEx
表达矩阵样本量很可观
- RSEM expected_count
(n=19,109)
UCSC Toil RNAseq Recompute - RSEM expected_count (DESeq2 standardized)
(n=19,039)
UCSC Toil RNAseq Recompute
RSEM expected_count output normalized using DESeq2 - RSEM fpkm
(n=19,131)
UCSC Toil RNAseq Recompute - RSEM norm_count
(n=19,120)
UCSC Toil RNAseq Recompute
TCGA TARGET GTEx gene expression by UCSC TOIL RNA-seq recompute - RSEM tpm
(n=19,131)
UCSC Toil RNAseq Recompute
PHENOTYPE
- TCGA GTEX main categories
(n=17,221)
UCSC Toil RNAseq Recompute - TCGA survival data
(n=10,496)
UCSC Toil RNAseq Recompute - TCGA TARGET GTEX selected phenotypes
(n=19,131)
UCSC Toil RNAseq Recompute
SOMATIC MUTATION (SNP AND INDEL)
- TCGA somatic mutations (Pan-cancer Atlas MC3 public version)
(n=8,463)
UCSC Toil RNAseq Recompute
TRANSCRIPT EXPRESSION RNASEQ
- RSEM expected_count
(n=19,109)
UCSC Toil RNAseq Recompute
TCGA TARGET GTEx transcript expression by RSEM using UCSC TOIL RNA-seq recompute - RSEM fpkm
(n=19,129)
UCSC Toil RNAseq Recompute
TCGA TARGET GTEx transcript expression by RSEM using UCSC TOIL RNA-seq recompute - RSEM isoform percentage
(n=19,131)
UCSC Toil RNAseq Recompute
TCGA TARGET GTEx transcript expression by RSEM using UCSC TOIL RNA-seq recompute - RSEM tpm
(n=19,131)
UCSC Toil RNAseq Recompute
TCGA TARGET GTEx transcript expression by RSEM using UCSC TOIL RNA-seq recompute
如果再进行去批次效应处理是否有错呢
你好进哥,我想要下载肝癌联合GTEX肝脏数据合并进行差异分析,使用UCSC下载,使用DEseq2进行差异分析,先下载了LIHC的count数据,GTEX是应该下载expected count还是nomal count数据进行分析,后面两种数据均是RSEM可以和LIHC直接合并然后处理批次效应吗?谢谢解答!
进哥请问TCGA TARGET GTEx队列里的RSEM expected_count数据集做基因差异分析,里面的TCGA跟GTEx样本要咋处理,还需要去除批次效应然后归一化吗,归一化的话用TMM或者GeTMM可以吗
这个数据集已经经过去批次化了,不需要再进行
https://www.jingege.wang/2023/05/24/3%e5%a4%a7%e6%95%b0%e6%8d%ae%e5%ba%93%e8%b6%852%e4%b8%87rna-seq%e6%95%b0%e6%8d%ae%e9%87%8d%e6%96%b0%e7%bb%9f%e4%b8%80%e5%a4%84%e7%90%86-%e5%85%b3%e4%ba%8etcga-gtex%e6%98%af%e5%90%a6/
你好,进哥哥
请问RSEM norm_count数据可否用limma包进行差异分析?SPSS等统计软件呢?
可以 说明数据来源 方法中
你好,进哥哥
我想请教一下,cohort: TCGA TARGET GTEx样本表达量数据中的RSEM norm_count数据是进行了哪种处理了呢?是否有去除批次效应,是否进行了标准化?我用这个数据进行差异分析的话是否还需要进一步处理?
你好,那个文章文献里有具体讲处理流程,总的来说用的python toil工具进行的整合,已经去除batch effect,也进行了normalization;差异分析不需要再进一步对数据处理
如果再进行去批次效应处理是否有错呢