写在前面:进哥本科生物技术专业,硕士博士都是卫生毒理学,目前博后仍然是卫生毒理学,为什么要搞这些数据库和分析工具呢?两个方面,其中最主要的可能是在基础研究上感觉到了迷茫和无力,不知道做了一篇完整的paper到底有什么实际意义,找不到成就感,想做一些更有意义和成就感的事;另一个就是我平时研究,包括本身的肺癌方向研究以及合作者其他肿瘤类型的研究,很多分析都是套路分析,就想着做一个分析组件。
目前已经完成的有三个数据库/R包/App:
1. 转录因子-靶基因预测工具TF Target Finder:https://mp.weixin.qq.com/s/gWvwI5Tx8e4IDZjpT8Fusg ;
2. 基于CPTAC数据库的癌症多组学(蛋白组/转录组/磷酸化蛋白组)分析套件PCAS:https://mp.weixin.qq.com/s/sa17MzmAOuulK1IS8foYoQ ;
3. 本文介绍的工具。
另外正在搭建的数据库是癌症预后数据库,涵盖生存分析相关的一系列功能,目前已完成代码部门,正在完善数据库,敬请期待!
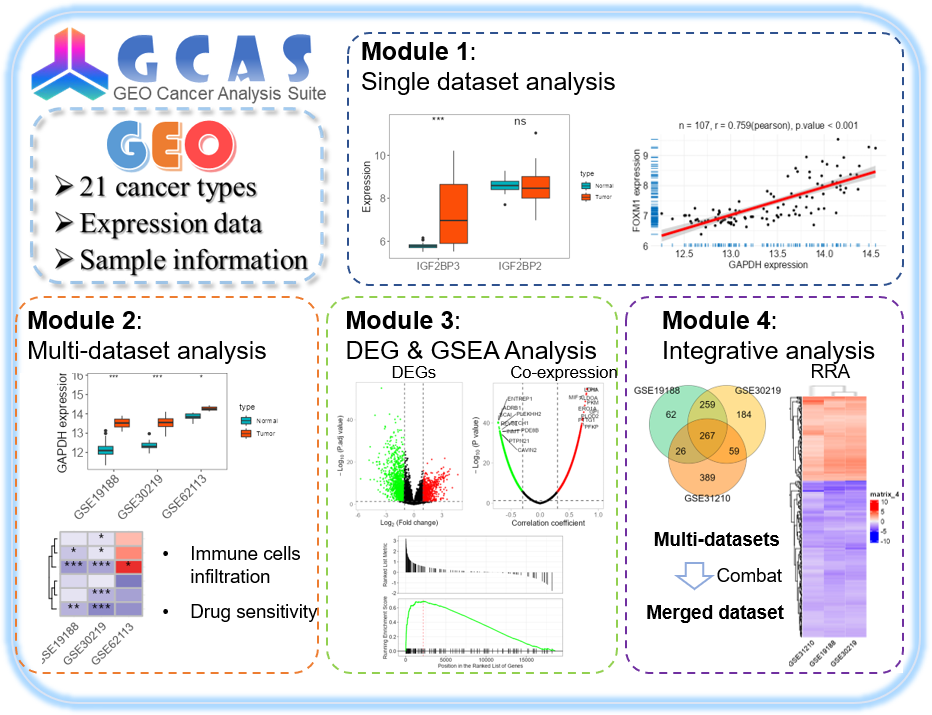
本文做简要介绍,关于GCAS的具体安装使用说明请参考:https://github.com/WangJin93/GCAS
以及B站视频演示:https://www.bilibili.com/video/BV1F19xY2EjS/
1. 简介
Title: GEO癌症分析套件(GCAS)
Version: 1.0.0
Author: Jin wang (Jin.wang93@outlook.com)
Maintainer: Jin wang (Jin.wang93@outlook.com)
Description: GEO癌症分析套件(GCAS)是一个多功能的R包,旨在分析和可视化癌症研究中的基因表达数据。GCAS允许对正常样本和肿瘤样本之间的基因表达进行比较、相关性分析、免疫浸润分析、差异表达分析、共表达分析和富集分析。它包含一个Shiny应用程序,用于交互式可视化,也可以直接在R环境中用于高级脚本编写。GCAS非常适合希望高效、有效地探索癌症基因组数据的研究人员、临床医生和生物信息学家。
Depends: R (>= 3.5.0)
Imports: RobustRankAggreg, VennDiagram, digest, dplyr, ggpubr, ggrepel, httr, jsonlite, meta, psych, shiny, stringr, sva, tibble, RColorBrewer, clusterProfiler, dplyr, ggrepel, grid, ggplot2
Encoding: UTF-8
URL: https://github.com/WangJin93/GCAS
Bug Reports: https://github.com/WangJin93/GCAS/issues
License: MIT License
2. 安装
remotes::install_github("WangJin93/GCAS") 3. 功能演示
描述
该函数用于从指定的数据集中检索特定基因的表达数据。
示例
单个数据集多个基因:
results <- get_expr_data(datasets = "GSE74706", genes = c("GAPDH","TNS1")) 单个基因多个数据集:
results <- get_expr_data(datasets = c("GSE62113","GSE74706"), genes = "GAPDH") 多基因多数据集:
results <- get_expr_data(datasets = c("GSE62113","GSE74706"), genes = c("SIRPA","CTLA4","TIGIT","LAG3","VSIR","LILRB2","SIGLEC7","HAVCR2","LILRB4","PDCD1","BTLA")) 描述
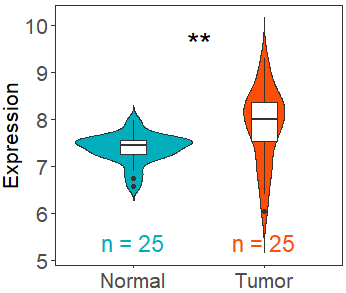
可视化GEO数据库中肿瘤组织和正常组织之间的mRNA表达数据的不同。
示例
df_single <- get_expr_data(datasets = "GSE27262",genes = c("TP53"))
viz_TvsN(df_single,df_type = "single")
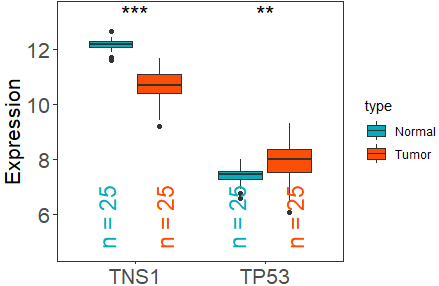
df_multi_gene <- get_expr_data(datasets = "GSE27262",genes = c("TP53","TNS1"))
viz_TvsN(df_multi_gene,df_type = "multi_gene",tumor_subtype ="LC")
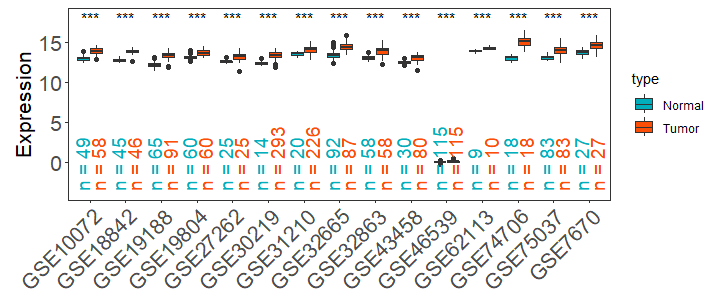
df_multi_set <- get_expr_data(datasets = c("GSE27262","GSE7670","GSE19188","GSE19804","GSE30219","GSE31210","GSE32665","GSE32863","GSE43458","GSE46539","GSE75037","GSE10072","GSE74706","GSE18842","GSE62113"), genes = "GAPDH")
viz_TvsN(df_multi_set,df_type = "multi_set")
描述
计算不同数据集中基因表达数据的摘要统计量(均值、标准差等)并进行假设检验(t 检验或 Wilcoxon 检验)。
示例
df <- get_expr_data(datasets = c("GSE27262","GSE7670","GSE19188","GSE19804","GSE30219","GSE31210","GSE32665","GSE32863","GSE43458","GSE46539","GSE75037","GSE10072","GSE74706","GSE18842","GSE62113"), genes = "GAPDH")
results <- data_summary(df, tumor_subtype = "LUAD")
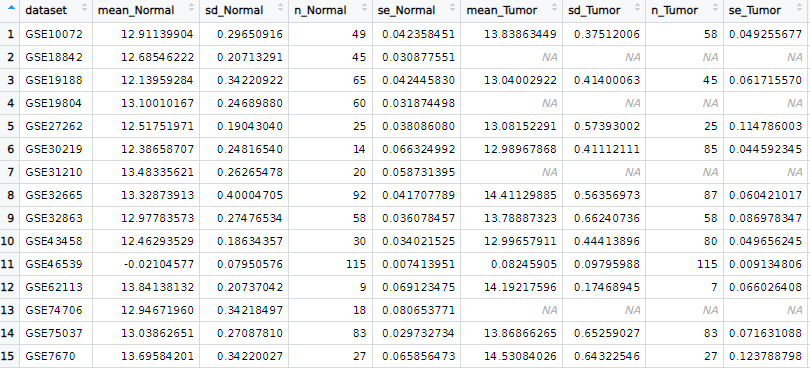
描述
绘制CPTAC数据集中肿瘤样本与正常样本之间差异表达基因(DEGs)的火山图。该功能对多个数据集进行荟萃分析,并生成森林图。同时,它还测试出版偏倚。
示例
df <- get_expr_data(datasets = c("GSE27262","GSE7670","GSE19188","GSE19804","GSE30219","GSE31210","GSE32665","GSE32863","GSE43458","GSE46539","GSE75037","GSE10072","GSE74706","GSE18842","GSE62113"), genes = "GAPDH")
results <- data_summary(df, tumor_subtype = "LUAD")
plot_meta_forest(results)
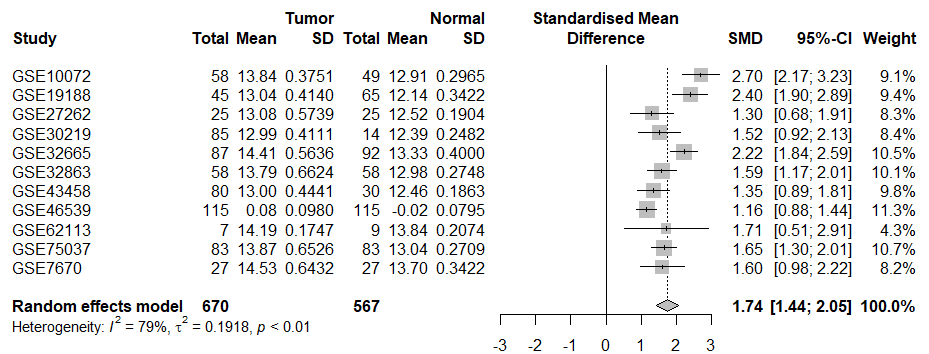
描述
此功能生成基因在不同数据集中的对数倍数变化(log fold change, logFC)热图。热图中包含基于p值的显著性注释。
示例
df <- get_expr_data(datasets = c("GSE27262","GSE7670","GSE19188","GSE19804","GSE30219","GSE31210","GSE32665","GSE32863","GSE43458","GSE46539","GSE75037","GSE10072","GSE74706","GSE18842","GSE62113"), genes = "GAPDH")
results <- data_summary(df, tumor_subtype = "LUAD")
heatmap <- plot_logFC_heatmap(results)
print(heatmap)
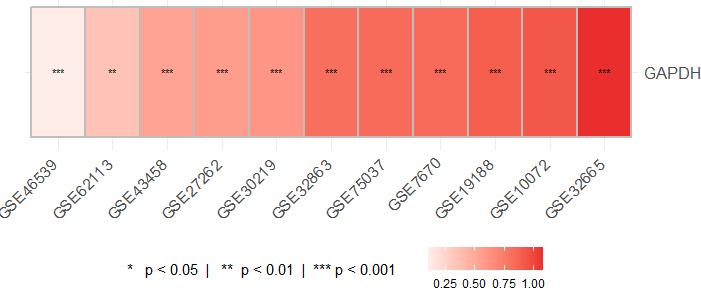
描述
此功能生成基因在不同数据集中的对数倍数变化(log fold change, logFC)散点图。散点图中包含基于p值的显著性注释。
示例
df <- get_expr_data(datasets = c("GSE27262","GSE7670","GSE19188","GSE19804","GSE30219","GSE31210","GSE32665","GSE32863","GSE43458","GSE46539","GSE75037","GSE10072","GSE74706","GSE18842","GSE62113"), genes = "GAPDH")
results <- data_summary(df, tumor_subtype = "LUAD")
scatter <- plot_logFC_scatter(results, logFC.cut = 0.5, colors = c("blue","grey20", "red"))
print(scatter)
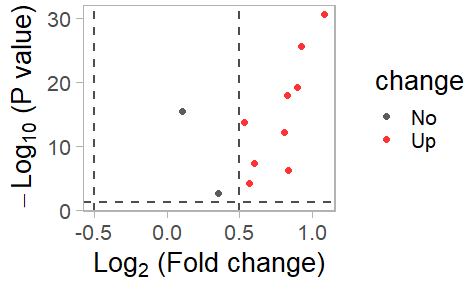
描述
对CPTAC数据库中的mRNA/蛋白质表达数据执行相关性分析。
示例
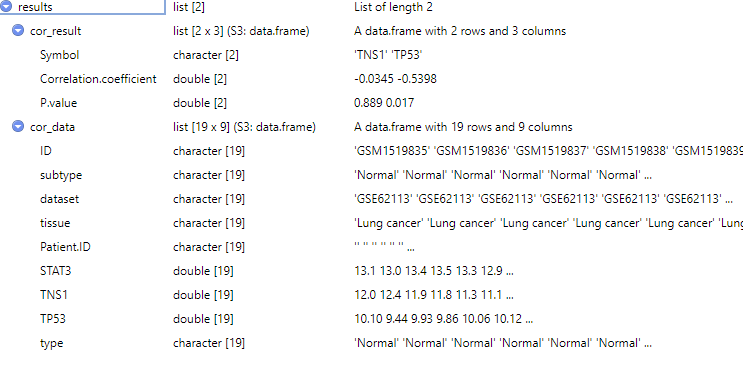
results <- cor_cancer_genelist(dataset = "GSE62113",
id1 = "STAT3",tumor_subtype = "LC",
id2 = c("TNS1", "TP53"),
sample_type = c("Tumor", "Normal"),
cor_method = "pearson")
描述
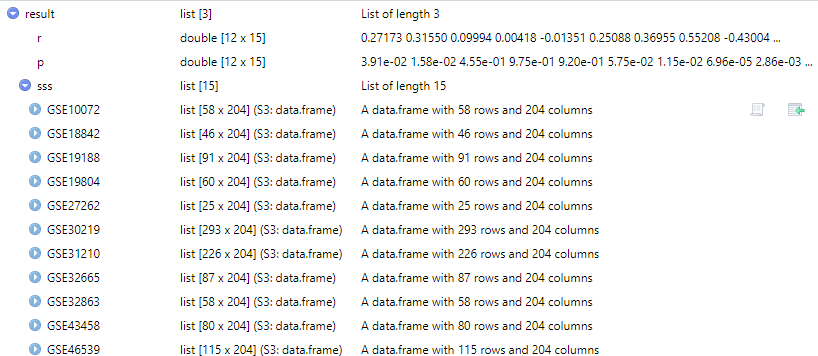
计算目标基因表达与抗肿瘤药物敏感性在多个数据集之间的相关性。
示例
dataset <- c("GSE27262","GSE7670","GSE19188","GSE19804","GSE30219","GSE31210",
"GSE32665","GSE32863","GSE43458","GSE46539","GSE75037","GSE10072",
"GSE74706","GSE18842","GSE62113")
df <- get_expr_data(genes = "TNS1", datasets = dataset)
result <- cor_gcas_drug(df, Target.pathway = c("Cell cycle"))
描述
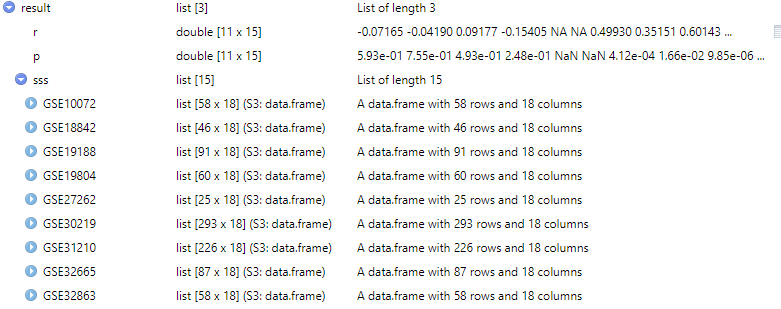
对多个数据集中的表达数据执行相关性分析。
示例
genelist <- c("SIRPA","CTLA4","TIGIT","LAG3","VSIR","LILRB2","SIGLEC7","HAVCR2","LILRB4","PDCD1","BTLA")
dataset <- c("GSE27262","GSE7670","GSE19188","GSE19804","GSE30219","GSE31210","GSE32665","GSE32863","GSE43458","GSE46539","GSE75037","GSE10072","GSE74706","GSE18842","GSE62113")
df <- get_expr_data(genes = "TNS1",datasets = dataset)
geneset_data <- get_expr_data(genes = genelist ,datasets = dataset)
result <- cor_gcas_genelist(df, geneset_data, sample_type = c("Tumor"))
描述
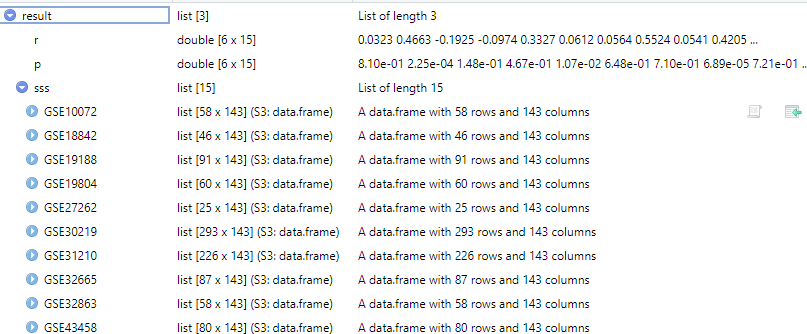
计算目标基因表达与免疫细胞浸润在多个数据集之间的相关性。
示例
dataset <- c("GSE27262", "GSE7670", "GSE19188", "GSE19804", "GSE30219",
"GSE31210", "GSE32665", "GSE32863", "GSE43458", "GSE46539",
"GSE75037", "GSE10072", "GSE74706", "GSE18842", "GSE62113")
df <- get_expr_data(genes = "TNS1", datasets = dataset)
result <- cor_gcas_TIL(df, cor_method = "spearman", TIL_type = "TIMER")
描述
使用基于 ggplot2 的热图展示相关性分析结果。
示例
上述相关性分析的所有结果均可以利用这个函数进行可视化,以免疫浸润结果为例:
viz_cor_heatmap(result$r, result$p) 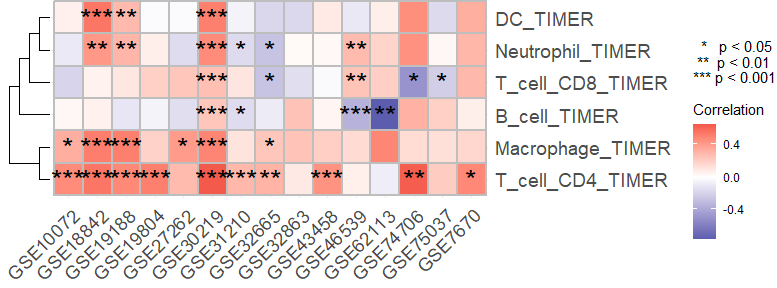
描述
绘制散点图,包含样本大小(n)、相关系数(r)和p值(p.Value)。
示例
viz_corplot(result$sss$GSE10072,"T_cell_CD4_TIMER","TNS1",x_lab = "") 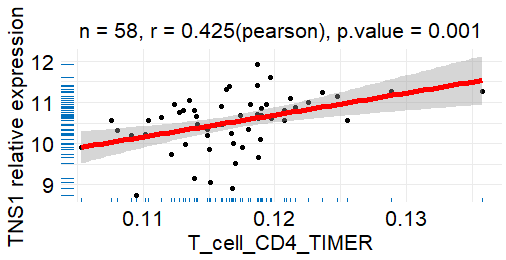
好了,本次介绍就到这儿,我太懒了,不想写了,明天再介绍其它高级功能。
工具目前正在投稿,等顺利录用之后我会更新在app和R包以及公众号和我的网站,先分享给需要的朋友,后续需要引用的时候再去这几个地方找一下。有建议请在github提交,谢谢啦!
Shiny APP:
在线App版本(人多会卡):https://jingle.shinyapps.io/gcas/
本地版本按照说明安装:https://github.com/WangJin93/GCAS
本地安装好R包后,运行 GCAS::GCAS_app()打开App
进哥,网站里面包含的这么多GEO数据是怎么找到的呀
嘿嘿,手动整理,文献检索,后续会持续更新