RNA pull-down 第一天
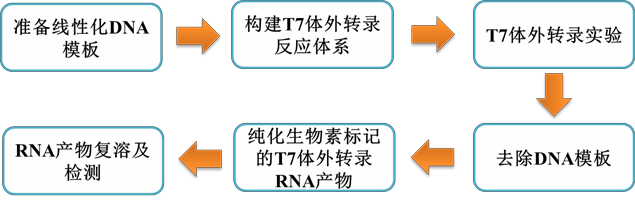
1. 线性化 DNA 模板的制备
本试剂盒(RiboTM RNAmax-T7 生物素标记转录试剂盒,RN:R11074.2)以含有 T7 启动子的线性化 DNA 作为模板。DNA 模板可使用线性化后的质粒,需避免线性化处理的 DNA 模板产生 粘性末端,如不可避免则需要进行平末端化处理。线性 DNA 模板有纯度要求,不可含有 RNase 污染,如有 RNase 污染,需去除 污染并重新回收。若模板来自 PCR 产物,则需要对 PCR 产物进行纯化,去除引物、引物二聚体及非特异性 DNA 扩增产物等杂质 后方可进行转录反应。
只有检验合格的线性化 DNA 模板才能与本试剂盒配套使用,质量好的线性化 DNA 模板才能转录制备出高质量的 RNA。故经 纯化的线性 DNA 模板需要进行严格的质控,可使用琼脂糖凝胶电泳或聚丙烯酰胺凝胶电泳进行质控,验证 DNA 模板线性化是否 完全,线性化片段大小是否正确,线性化的 DNA 模板片段条带是否单一等。
2. T7 体外转录反应体系的配置与转录实验操作
按下表配置体外转录反应体系:
表2 体外转录反应体系
|
序号 |
T7 体外转录反应体系 |
用量 |
|
1 |
线性化 DNA template (0.1~0.2μg ) |
< 8μl |
|
2 |
T7 Enzyme mix |
2μl |
|
3 |
T7 Reaction Buffer(5X) |
4μl |
|
4 |
T7 RNA Labeling Mix |
6μl |
|
5 |
RNase Free H2O |
Add to 20μl |
用移液器轻轻吸打混匀反应体系(请勿剧烈震荡混匀),瞬时离心以确保溶液全部汇集在管底,置于热循环仪上 37℃孵育反 应 2~4h(反应时间可根据合成 RNA 长度大小进行适当调整优化),反应结束后将体系置于冰上,建议尽快进行下一步纯化,不 建议将体系冻存。
3. 去除 DNA 模板的操作
向上一步的反应体系中加入 1μl DNase I(1U/μl),并用移液器混合均匀,瞬时离心使溶液全部汇集于管底,置于热循环仪 上 37℃孵育反应 20min,以彻底消除体系中的 DNA 模板,可根据 DNA 模板长度适当优化反应时间。
4. T7 体外转录 RNA 产物的纯化
按下表所述配制 RNA 产物的纯化体系:
表3 RNA 产物纯化体系
|
序号 |
RNA 产物的纯化体系 |
用量 |
|
1 |
去除模板后的转录产物 |
21μl |
|
2 |
RNase Free H2O |
67μl |
|
3 |
Purification Assistant A |
10μl |
|
4 |
Purification Assistant B |
2μl |
*建议使用灭菌后的 1.5ml 低吸附离心管进行纯化
轻轻混合均匀后加入预冷无水乙醇 300μl,轻轻混合均匀后放置于-20℃冰箱中两个小时或过夜沉淀,若放置于-80℃的冰箱中,则至 少需要沉淀 1 小时以上。
沉淀后,使用低温冷冻离心机在 4℃、13000rpm 条件下离心 30min,离心后可明显看到 RNA 沉淀。弃去上清后加入 1ml 预冷 70%
乙醇,颠倒离心管洗涤沉淀。建议重复洗涤步骤一次。
最后于低温冷冻离心机在 4℃、13000rpm 条件下离心 10min,弃去上清后(尽量把剩余乙醇洗液全部去除)干燥晾干 RNA
沉淀至透明。
5. RNA 产物的复溶及质检
加入 30~50μl RNase Free Water 或 1 X TE 溶液溶解晾干后的 RNA 沉淀,室温放置 5min 直至 RNA 沉淀充分溶解。使用微量分 光光度计(如 Nanodrop)或 Qubit 等对 RNA 的浓度及纯度进行检测,使用琼脂糖凝胶电泳或者 Agilent 2100、Agilent2200 等仪器 检测确认 RNA 的完整性。
RNA pull-down 第二天
1、前期准备
·DTT,蛋白三联室温溶解;
·streptavidinmagnetic beads;(S1420, New England Biolabs)
·RNase Inhibitor;
·细胞,10cm大皿若干,长至约80~90%密度;

c、1M DTT:
DTT 溶液用0.01M 乙酸钠(PH=5.2)配制,1g DTT粉剂加入6.48ml 乙酸钠溶液中,溶解后过滤,分装后冻存于-20℃;
d、5M NaCl 50ml:
14.61g NaCl粉末加入50ml DEPC水中;
e、PBS(DEPC水配制), 250ml。
2、RNA抽提
a)将沉淀过夜的RNA取出,4℃,最大转速离心30min,离心后可见管底的白色沉淀,及为RNA;
b)小心移取上清,用1ml 70%乙醇洗涤,8000rpm离心10min,之后吸净管内液体,空气中晾干RNA,若乙醇未挥发完全,将影响下游实验效率;
c)每管加20μl nuclease free-water溶解RNA,之后将RNA放于PCR仪中,95℃,5min使其变性,之后立即置于冰上,备用。
3、细胞裂解
a)弃去培养基,用预冷的PBS洗3次,吸去上清;
b)每个大皿中加入200μl cell lysis buffer A (加蛋白三联);
c)用细胞刮刀刮下细胞,收集至EP管,液氮中三冻三融;
d)离心,4℃,3000rpm,离心10min;
e)转移上清至新的EP管中,置于冰上;可加200U/mlRNase Inhibitor;
f)取10-20μl左右上清至新管中,作为实验input组。
4、magneticbeads 预处理
a)涡旋混匀beads;
b)每管取400μl beads(如本次实验中,control RNA及目的RNA各1管),置于磁力架上,吸去上清;
c)用800μl 1X binding & washing buffer ,洗3次,吸去上清;
5、RNA与beads结合
a)向上一步的beads中加入400μl 2Xbinding & washing buffer;
b)向上一步中加入20μl RNA ,再补380μl DEPC水,使其终体积为800μl;
c)室温慢速旋转20min,使beads与RNA充分结合;
d)将上一步的管子置于磁力架上,吸去上清;
e)用800μl 1X binding & washing buffer(含RNaseInhibitor, 1U/ μl),洗3次,吸去上清;
f)用celllysis buffer A洗一次beads,吸去上清,注意不要让beads干掉。
6、RNA与蛋白相互作用
a)向上一步已处理好的beads中每管加入500μl细胞裂解液(第3步);同时向每管加入RNase Inhibitor, 1U/ μl;
b)4℃慢速旋转2h,使beads与细胞裂解液充分结合;
c)磁力架上吸去上清,用400 μl新鲜配制的celllysis buffer A(+RNase Inhibitor+蛋白三联),洗5次beads;
d)吸去上清,用25μl预冷的0.1% SDS溶液重悬beads,加6.25μl的5X蛋白上样缓冲液,100℃金属浴煮10min.之后立即置于冰上5min,再置于磁力架上吸取上清至新的EP管中,准备蛋白上样。
7、蛋白的检测
可采用western blot或质谱继续下游蛋白的验证。
进哥,“含有 T7 启动子的线性化 DNA 作为模板”,这一步是如何设计的有无教程。
你好,请问RNA很长的话,我要怎么构建DNA模板呢?
您好,看到有RiboTM RNAmax-T7 生物素标记转录试剂盒下面的推荐文章里都是直接加入相应质量的RNA与细胞裂解液孵育后在与磁珠一起孵育1h,我看您这里介绍的方法是先让RNA与磁珠结合后在与细胞裂解液孵育,这两种方法具体有哪些不同呢?另外就是binding&wash buffer还有cell lysis bufferA 都是自己配的吗?后者与RIRA这些强裂解液的区别是什么呢?希望能加上您的微信跟您讨论。
不好意思 最近太忙,网站留言没时间回复,微信上讨论
请问进哥,RNA抽提之后的95℃变性是必需的吗?若不进行变性步骤,是否会影响RNA和目的蛋白的结合效率?
您好,变性的目的是把一些体外转录形成的二级结构打开,可能有些二级结构会抑制RNA和蛋白的结合,我觉得嘛,不变性也可以的,毕竟正常细胞里面结合之前也没有变性的。。。
您好,我想问一下,做RNA-RNA pull down也可以使用RiboTM RNAmax-T7 生物素标记转录试剂盒吗?还有就是赛默飞试剂盒好像是末端标记,是不是和这个不一样?
当然可以用,前面原理步骤都一样
thermo试剂盒你指的什么不一样?如果用的链霉亲和素欧联的磁珠,都是需要标记biotin,不管那个牌子,目的一样,可能操作方法有差异
谢谢解答,赛默飞的试剂盒是体外转录后,在RNA产物末端加上标记,我看RiboTM RNAmax-T7 生物素标记转录试剂盒是可以直接体外转录出带生物素标记的RNA,我以为是针对不同的下游实验。
探针可以直接加热吗,需不需要加structure buffer
这个不是很清楚 不过我觉得是没有问题的
进哥~ 想请教一下~ 我是公司合成的生物素标记RNA~ 单位是nmol~ 请问你这里加的20ulRNA大概是多少ug呀~ 我看赛默飞的pulldown试剂盒用的是50pmolRNA和50ul的磁珠,换算到我的200bp的RNA 大概是2.5ug~
你好,请问是5、RNA与beads结合这一步的RNA用量吗?20ul是体外转录获得的RNA探针,上述步骤就是用20ul复溶的,全部加入就好,量就是一次体外转录的量,定量的话也没有必要,正常情况量是足够的
如果是其它问题 直接加微信讨论吧
请问有没有R studio软件呀 谢谢
R studio是免费软件哇 直接官网下载桌面版就好
宝藏博主!
谢谢,欢迎一起探讨