第一部分:设计shRNA序列
首先打开网站:http://rnaidesigner.thermofisher.com/rnaiexpress/setOption.do?designOption=shrna&pid=-9122351712654529539,在Target Design Options处选中shRNA,以Fli1为例,输入Accession number或Nucleotide sequence,其他条件不变:
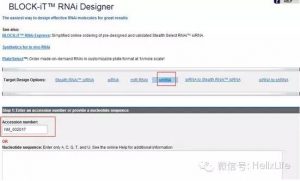
下拉到底点击RNAi Design,然后出现推荐的10条靶点序列:

根据Rank评星,选择其中需要的(进哥哥一般选择4条,不同位置各一条),点击Design shRNA Oligos:

然后在Default Loop Sequence选择合适的序列(进哥哥用的载体是pGreenPuro,不需要这个序列,就默认了,后面合成引物的时候要去掉的),在Custom Loop Sequence处输入CTCGAG,点击Design:

得到shRNA
需要特别提醒,如果要构建到慢病毒载体上,合成出去的shRNA引物会根据载体有些不一样,sigma用的是pLKO.1载体,小编常用SBI的pGreenPuro(CMV) 载体,两端的酶切位点是BamHI/EcoRI,设计出去的引物最终是这样的(不同shRNA替换中间红色部分):
Sense:GATCCCCCTTCTGACATCTCCTACATCTCGAGATGTAGGAGATGTCAGAAGGGTTTTTG
Anti-sense: AATTCAAAAACCCTTCTGACATCTCCTACATCTCGAGATGTAGGAGATGTCAGAAGGGG
还可以使用GPP这个工具设计:
第二部分:慢病毒载体构建(以pGreen-Puro)
步骤1 :退火寡核苷酸链
根据我们的经验,脱盐纯化的寡核苷酸足以满足连接和克隆需要。但是不同供应商提供的引物纯度差别很大,如果您连接和克隆遇到困难,可以考虑换用PAGE纯化或HPLC纯化的引物,或者使用其他供应商提供的引物。
用水将寡核苷酸稀释为100μM。按以下体系配制退火反应体系:
l 正义寡核苷酸(100μM)2μl
l 反义寡核苷酸(100μM)2μl
l 10X T4 DNA连接酶缓冲液 1μl
l 加水15μl补足20μl
退火寡核苷酸链
在PCR仪或沸水烧杯中95°C孵育4分钟。如果使用PCR仪,将样品在70°C孵育10分钟,然后在数小时内缓慢冷却至室温。如果使用烧杯水,请将烧杯从火焰中取出,并让水冷却至室温。这将需要几个小时,但对于寡核苷酸退火冷却发生缓慢是很重要的。
步骤2 :酶切载体
用EcoR I和BamH I双酶切2μg载体。酶切方法和体系参照您购买的内切酶说明书或者按照您的实验室习惯的方法进行。
对线性化的载体进行去磷酸化处理是没有必要的。充分酶切后的载体具有不匹配的末端,一般不会发生载体自连。
步骤3 :连接载体
用水将退火后寡核苷酸稀释100倍备用。按照以下体系配制连接反应体系:
l T4 DNA 连接酶 5U
l 线性化载体 2μl
l 稀释后寡核苷酸 2μl
l 10×连接酶Buffer 1μl
l 加水补足 10μl
接连反应条件和时间参照您购买的连接酶说明书进行。
为了减少空载体自连,连接反应完成后,在连接反应体系中加入1μl BglII,37℃反应30min,切割连接上的空载体。(选做)
步骤4 :转化大肠杆菌感受态
用连接后产物转化大肠杆菌感受态细胞。市场上常见的大肠杆菌感受态细胞(例如Top10,DH5α)均可以使用,您也可以按照分子克隆中的方法自己制备感受态细胞。转化方法按照供应商的说明书或者您实验室中常用的方法进行。
在氨苄抗性的琼脂平板上37℃培养转化后细菌,大约14-16小时后,平板上出现单个细菌菌落。挑取多个菌落至氨苄抗性的培养基中培养后进行鉴定。
步骤5 :转染细胞
pRI系列载体可用常用的转染方法进行细胞转染。包括:磷酸钙转染、脂质体转染、电穿孔转染等。您可以购买市售的商品化转染试剂并且参照供应商说明书进行细胞转染实验。
步骤6 :观测GFP 荧光和稳定表达细胞系筛选
如果细胞转染成功,并且您使用了带GFP的载体,根据细胞生长速度的不同,在转染后3-5天能在荧光显微镜下观察到细胞发出绿色荧光。请注意,绿色荧光蛋白的表达表明转染细胞成功,不能说明RNA干扰实验成功。
如果您使用了带有真核抗性标签如Neo-R的载体,这时您可以加入合适浓度的相应药物如G418进行稳定表达细胞株的筛选。
步骤7 :检测RNA 干扰效率
您可以在蛋白水平或mRNA水平检测RNA干扰效率。一般情况下蛋白的表达变化与mRNA水平的表达变化一致, 也有少数情况下mRNA表达水平变化不及蛋白表达下降明显。
为检测蛋白表达情况,您可以使用Western-Blot。
为检测mRNA表达情况,您可以使用逆转录+Real-time PCR或Northern-Blot。
具体检测方法请参照分子克隆使用指南或者您实验室的常用实验方法。
进哥哥您好!一直有个疑问,我使用MERCK网站查找Target序列,有些序列并不是G开头的,需要在前面加上一个G吗,原理是什么呢?期待您的回复。
您好,shRNA退火后与酶切后的载体连接,但是转化后无菌落。我怀疑是shRNA有发夹结构,退火过程中正确的双链shRNA较少,您认为有可能是这个原因吗,如果是这个原因,怎么解决呢?公司可以合成双链shRNADNA片段吗。
王进老师,您好,我遇到的问题是测序失败或者是测序结果对比之后连接上的序列不对,请问是哪里出问题呢
你好,根据该网站设计,我的shRNA序列只有53bp,因为前后也没有酶切位点,因此我选择环p,但是环p我采用各种酶都p不出来,请问可以帮我解答一下吗
为啥在5‘端加C末尾加G,有点不明白,我在GPP网站设计的只有末尾加G
进哥,这个网站现在进去之后,点设计没有设计结果,有其它进去方法吗?
你好:我有以下疑问:载体上的sense和antisense可不可以不一样长。我的需求是现在体内得到不是平末端的发卡结构(环不要被切掉),可以实现吗
你好!请问我按照步骤输入NCBI上查取的编号输进去,设计ShRNA没有结果显示是怎么回事呢?
您好,我有个问题,如果我们设计的靶基因的序列首位不是从G开始的,那么我们合成退火序列的时候需要在酶切位点后手动添加吗?
请问为什么赛默飞那个设计sh的点进去后出现了乱码,有什么解决办法吗?
请问为什么引物的酶切位点分开了啊
老师,你好,我想问下这个干扰体系可以用在昆虫的基因敲降吗?在昆虫中的干扰效率如何?
进哥好~构建RNAi筛选库,在shRNA两端加上引物序列形成Oligo pool,您知道具体如何设计及原理嘛?
请问你的问题解决了么,最近也遇到了这个问题,测序结果很多是单个碱基错配或者头尾有缺失,而且测序很短,公司说是测不过去。
进哥你好,请问这个设计引物序列的网站除了人之外可以设计鱼的吗?这两个载体都可以用于构建鱼的shRNA载体吗?应该没有物种的区别吧……
您好,请问对从菌液中提取的shRNA质粒进行双酶切验证时,跑胶出现三条条带可能是因为什么呢?
你好,我使用这个网站设计输入我的lncRNA的orf之后显示No results are found within the unique regions of the input sequence. Change minimum and maximum GC% and try again.我尝试着调整了GC%之后依然没有,怎么解决呢。
shRNA的设计 ——GPP Web Portal – 王进的个人网站
https://www.jingege.wang/2021/09/06/shrna%e7%9a%84%e8%ae%be%e8%ae%a1-gpp-web-portal/
那就试试其它网站吧,参数有点严格,得不到结果
您好,我已知si序列,序列后面有个dtdt悬垂,我用si序列的方法构建sh载体,不知道要不要加上悬垂dtdt,希望能收到解答,谢谢您
不用的,shRNA本身可以维持一定的稳定性。当然加上会不会更好我也没有比较,建议做的时候同时比较一下,也没有很麻烦
您好,请问一下基因的编号怎么搜索啊
进哥好~请问您知道两个shRNA构建到同一个慢病毒载体怎么做嘛 中间需要用什么linker连接呢~谢谢~
因为shRNA比较短,可以直接用各自的的转录元件,主要就是U6启动子了,明白,就是两个启动子,各自负责shRNA
但是这样引物怎么设计呢 U6不是在载体 酶切后把我的shrna塞到U6后面这样吗
进哥哥好,我想问几个问题:
1.shRNA慢病毒转染细胞,需要控制血清,购买专门的培养基(例如opti)嘛?
2.慢病毒转染和病毒侵染的区别是什么呀?
3.我转染细胞的操作是细胞铺板过夜后,直接加入病毒,转染6-8小时后换新的培养基,完全培养基还是常用的10%FBS,1%双抗,和基础培养基构成的。但是48h后在镜下看荧光表达不高,这是为什么呢?
4.WB结果显示目的基因敲低,但是下游主要的信号通路并没有发生变化,这是什么原因呢,是shRNA设计序列不对(但是目的基因确实敲低了),还是有其他补偿机制(但是文献中下游通路都发生了改变)呢?我需要再加大病毒浓度嘛?
收到您的疑问 方便的话直接加微信讨论吧 我的简历里面有手机号微信号
老师,第4点问题跟您是一样的,是否需要再构建质粒去做功能实验?
您好进哥哥,我想问下步骤4中的“挑取多个菌落至氨苄抗性的培养基中培养后进行鉴定”,是指提质粒测序进行鉴定吗?谢谢您的分享
可以 PCR初筛 测序确定
进哥哥你好,请问测序结果有很多不匹配是什么原因呢
什么意思?sanger测序首尾会有几十bp不匹配的,中间一般不会,如果质粒纯的话,不应该,可能是不是有其它质粒污染
请问你的问题解决了么,最近也遇到了这个问题,测序结果很多是单个碱基错配或者头尾有缺失,而且测序很短,公司说是测不过去
你好,请教一下前面关于蛋白的转录本怎么选择呢
你好 不同转录本编码的蛋白不一样 或者有些不编码蛋白 根据你所研究的蛋白选择,查看对应转录本的蛋白产物 建议使用Ensembl数据库查看
厉害,生物强人。来之同为生物人的评论和点赞
谢谢 一起学习讨论
进哥你好 测序完之后shrna那段有几个碱基不对 不知道是什么原因