有这样的使用场景么?
1.已经确定研究的基因,但是想探索他潜在的功能,可以通过跟这个基因表达最相关的基因来反推他的功能,这种方法在英语中称为guilt of association,协同犯罪。
2.我们的注释方法依赖于TCGA大样本,既然他可以注释基因,那么任何跟肿瘤相关的基因都可以被注释,包括长链非编码RNA。
GSEA需要的gene set是现成的没有问题,但是genelist没有,这里我们可以把所有基因跟单个基因的相关性系数当做LogFC,有正有负,就解决了geneList的问题。这个想法不是我的,是我的一个学员的,不过他要解决的是microRNA把基因的问题。
下面来实战一下:
1.首先加载数据
这个数据是我下载了TPM数据,然后提取出乳腺癌的数据得来的。
load(file = 'BRCA_mRNA_exprSet.Rdata')
exprSet <- mRNA_exprSet
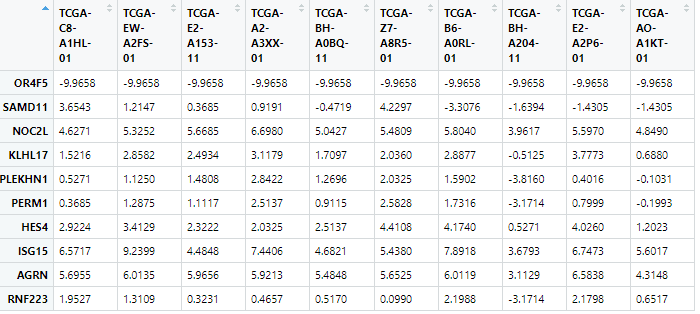
test <- exprSet[1:10,1:10]
2.写一个函数批量计算相关性
这个函数只要输入一个基因,他就会批量计算这个基因跟其他编码基因的相关
性,返回相关性系数和p值。
###对于有缺失值的基因,有效样本小于4会报错
batch_cor <- function(gene){
y <- as.numeric(exprSet[gene,])
rownames <- rownames(exprSet)
do.call(rbind,future_lapply(rownames, function(x){
dd <- cor.test(as.numeric(exprSet[x,]),y,type='spearman')
data.frame(gene=gene,mRNAs=x,cor=dd$estimate,p.value=dd$p.value )
}))
}
###这是修改的代码 加一个判断 样本量<10的就不要了吧
batch_cor <- function(gene){
rownames <- rownames(exprSet)
do.call(rbind,future_lapply(rownames, function(x){
xy <- exprSet[c(gene,x),]
xy <- t(xy) %>% na.omit() %>% as.data.frame()
if (nrow(xy)>10){
dd <- cor.test(as.numeric(xy[,1]),as.numeric(xy[,2]),type='spearman')
data.frame(gene=gene,mRNAs=x,cor=dd$estimate,p.value=dd$p.value )
}
}))
}
3.并行化运行函数
以PCDC1这个基因为例
library(future.apply)
plan(multiprocess)
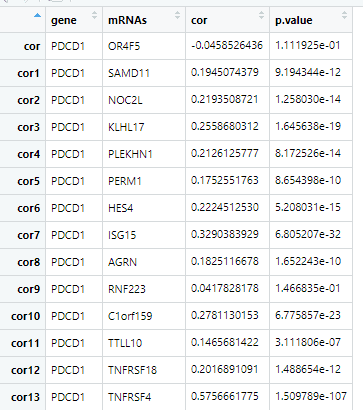
system.time(dd <- batch_cor('PDCD1'))
这是返回的结果
4.制作genelist
gene <- dd$mRNAs## 转换
library(clusterProfiler)
gene = bitr(gene, fromType='SYMBOL', toType='ENTREZID', OrgDb='org.Hs.eg.db')## 去重
gene <- dplyr::distinct(gene,SYMBOL,.keep_all=TRUE)
gene_df <- data.frame(logFC=dd$cor,
SYMBOL = dd$mRNAs)
gene_df <- merge(gene_df,gene,by='SYMBOL')
## geneList 三部曲
## 1.获取基因logFC
geneList <- gene_df$logFC
## 2.命名
names(geneList) = gene_df$ENTREZID
## 3.排序很重要
geneList = sort(geneList, decreasing = TRUE)
5.运行GSEA分析
从GESA(https://www.gsea-msigdb.org/gsea/downloads.jsp)的官网上,下载一个gmt文件
library(clusterProfiler)
## 读入hallmarks gene set,从哪来?
hallmarks <- read.gmt('h.all.v6.2.entrez.gmt')
# 需要网络
y <- GSEA(geneList,TERM2GENE =hallmarks)
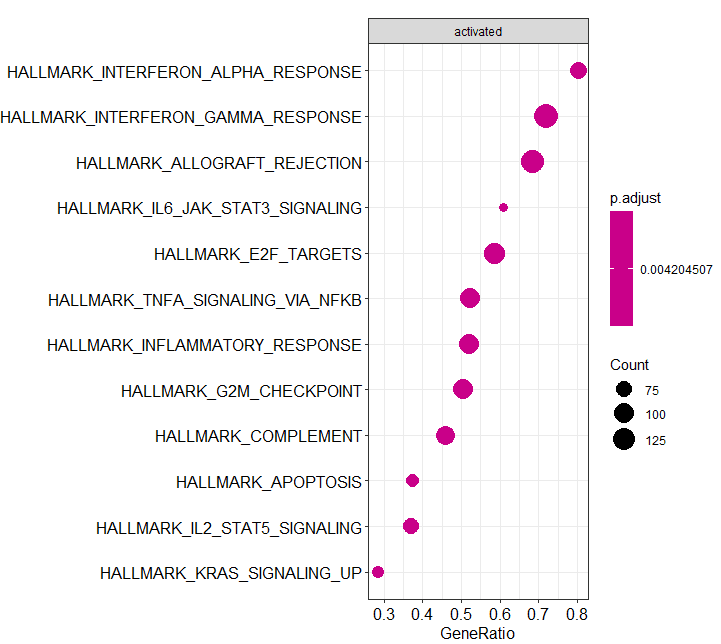
作图看整体分布
### 看整体分布library(ggplot2)
dotplot(y,showCategory=12,split='.sign')+facet_grid(~.sign)
本次结果中全是激活的
6.特定通路作图
yd <- data.frame(y)
library(enrichplot)
gseaplot2(y,'HALLMARK_INTERFERON_ALPHA_RESPONSE',color = 'red',pvalue_table = T)
PCDC1跟阿拉法干扰素正相关,这个事情没什么好说的吧。

如需要批量进行几个基因的分析,可以包进一个函数会更方便。
好了,我们又掌握了一个特别强悍,实用的技能。我是进哥哥,有问题随时留言讨论。
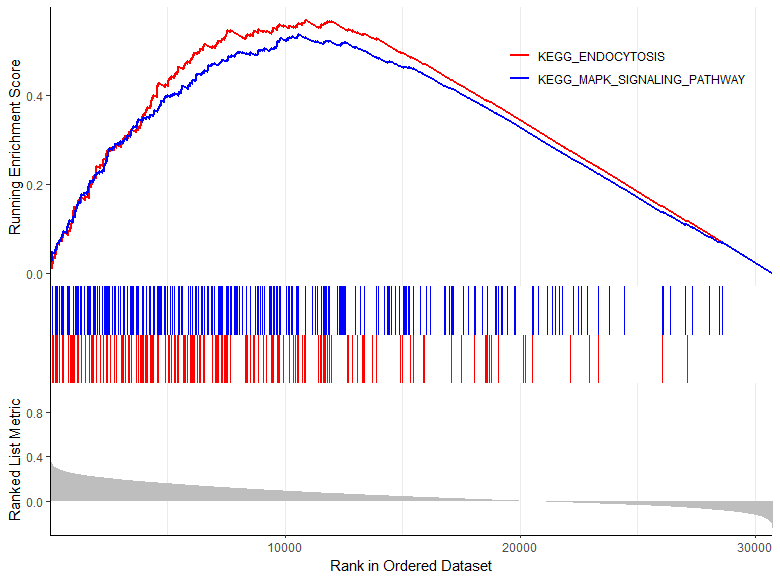
##对于多个通路绘制在一起:::
pathway=c("KEGG_ENDOCYTOSIS","KEGG_MAPK_SIGNALING_PATHWAY")
gseaplot2(y,pathway,color = c('red',"blue"),pvalue_table = F)
你好,王老师,单基因GSEA富集的时候可以用count值吗?
您好,请问按照这样进行分析后,结果中NES为正值是代表与目标基因正相关的基因集富集在此通路上吗,能由此推测目标基因是激活该通路吗?还是只是说明该基因参与此通路,发挥的作用是激活还是抑制有待后续研究?
进哥你好,本人纯生信小白,我从XENA上下载了肿瘤的TPM数据,请问怎么把基因名注释进去,弄成您这样的示例数据?
请问是将得到所有相关性基因进行GSEA分析的吗,不需要筛选Pvalue值<0.05的基因,做GSEA分析吗
您好,可以去掉plan(multiprocess)这一步吗
txt文件怎么转换为Rdata呢
您好,为什么GSEA的点图展示中所有的结果P值都是一样的呀,旁边的颜色图例应该是连续渐变的呀
进哥,这个是用TPM,还是对数化后的TPM呀?
进哥,我遇到的问题是
1、做相关性分析的时候,要不要对结果进行P值的筛选,在进行后续的富集,
2、某因子可能抑制某条通路下游通路(比如某因子抑制NF-kB信号通路的下游P65的磷酸化),做出来NES图是不是应该负相关?还是没有变化(因为他是蛋白修饰发生改变而不是蛋白表达量发生改变)?
3、多个癌肿中分析信号通路取交集得到大部分影响细胞周期中的生物学功能是否提示影响这些肿瘤增殖?
Nature. 2021 Jul;595(7866):309-314
补充图3
load(file = ‘BRCA_mRNA_exprSet.Rdata’)#这个file必须是Rdata格式吗,没看懂这块,大佬能提供下这块的上一步代码操作码
exprSet <- mRNA_exprSet#这里面mRNA_exprSet在前面没出现过,可以这么操作吗
基于相关性分析进行GSEA是否有文献参考呢 我查了没查到 不知道是不是在方法里写清楚就行?
写清楚方法吧,这个常规做法,按照相关性系数作为权重进行GSEA
不太能够理解,以示例图为例子的话,这种方法是说明相关度高的基因在该通路中起贡献作用,相关度低的基因起抑制作用,从而推测该基因与贡献相关,这种方法也可以应用于go富集分析么,
这个方法并不能说明activate还是inhibit作用,只能表示对该通路的贡献。无非就是得到一个按照特定权重排序的基因集,这里是相关性系数,差异分析是logFC
其他分析也一样,GO、Cluster等等
好的好的,感谢解惑
基于相关性分析进行GSEA是否有文献参考呢 我查了没查到 不知道是不是在方法里写清楚就行?
您好,请问TPM数据是只需要肿瘤患者的数据还是要肿瘤+正常患者的数据呢?
因为基于相关性分析嘛 其实加上正常组织未尝不可,你方法中写清楚就好
好的!谢谢!
您好,请问为什么运行到system.time(dd <- batch_cor('PDCD1'))这一步就变得特别慢,甚至一个小时都不出结果?
你的数据很大?还是电脑配置不够?并行运行需要根据电脑配置修改一些参数 ,不然很卡很慢
或者直接用for循环进行 不并行处理
数据倒也不大以前一直可以运行的,最后用for循环解决啦!感谢!
error in if(abs(max.ES)>abs(min.ES)){:missing value where TRUE/FALSE needed,请问老师,这个是什么意思呢,最后的出图出不来。望老师帮帮我。
这样看不出来诶。。。确定你所要画的通路在富集结果里面吗 并且拼写正确?可以加我微信具体讨论
我刚刚也留言碰到同样的问题 找了GitHub也有人有相同疑问 我在另一个帖子下面留言了 o(╥﹏╥)o
您好,本人也困于此问题,请问您于王博交流后,此问题如何处理的呢。烦请告知,多有打扰,谢谢~
加我微信吧 得一步步运行看看什么问题
请问这个问题解决了吗,我也遇到了同样的问题
不清楚指的那个问题?都可以解决 如果还没解决 可以加我微信 我看看
您好,可以提供一下PTM吗?想试试
您好 发给你数据太大,这个不一定需要TPM数据,只要是任意标准化的表达矩阵都可以,你可以从XENA上下载任意肿瘤的表达矩阵进行分析,你先试试 有问题加我微信讨论
在进哥的指导下,运行成功,再次感谢进哥!
您好!非常感谢您提供便捷的学习资料。想问下您:我在运行“plan(multiprocess);system.time(dd <- batch_cor('PDCD1'))”时,显示“ Error: ‘node$session_info$process$pid == pid’ is not TRUE ” ,这个问题应该怎么解决。谢谢
你好,这个得看你的数据,要不你加我微信详细讨论,我的简历最下面有微信
请问要单基因的gsea图包含多条通路怎么做呢,谢谢!
哈哈,晚一点出个教程
对于clusterprofiler包的结果,直接绘制时添加多个通路即可,文中最后已补充 请参考
你好,王老师,多个通路的颜色没办法自己绘制,是不是要加个颜色的代码。