以肺腺癌数据(TCGA-LUAD)为例,为了用TCGA结直肠癌数据做分析,我们首先要先整理出该癌症的基因表达矩阵。(也有一些数据库提供整理好的TCGA癌症数据,如UCSC xena数据库对TCGA数据进行了整理,可直接下载表达矩阵和临床数据用于研究)
进入GDC data portal–>Respository栏目,勾选下面选项:(注意,TCGA更新后的Workflow Type一栏只有STAR – Counts,即将原来的HTSeq-Counts、HTSeq-FPKM、HTSeq-FPKM-UQ数据都放入了一个文件中)
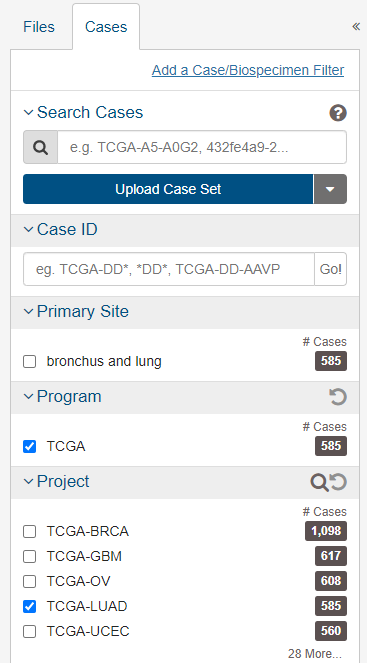
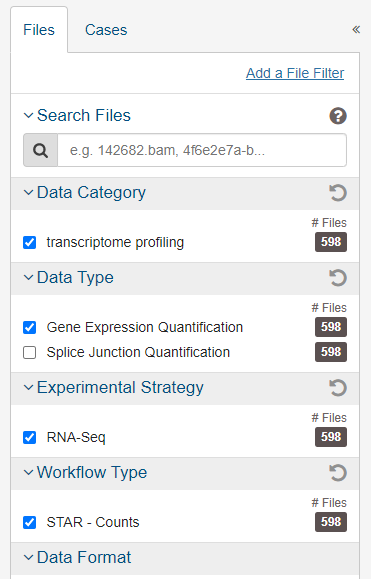
对筛选到的文件,可一键全部添加到cart或手动添加到cart:
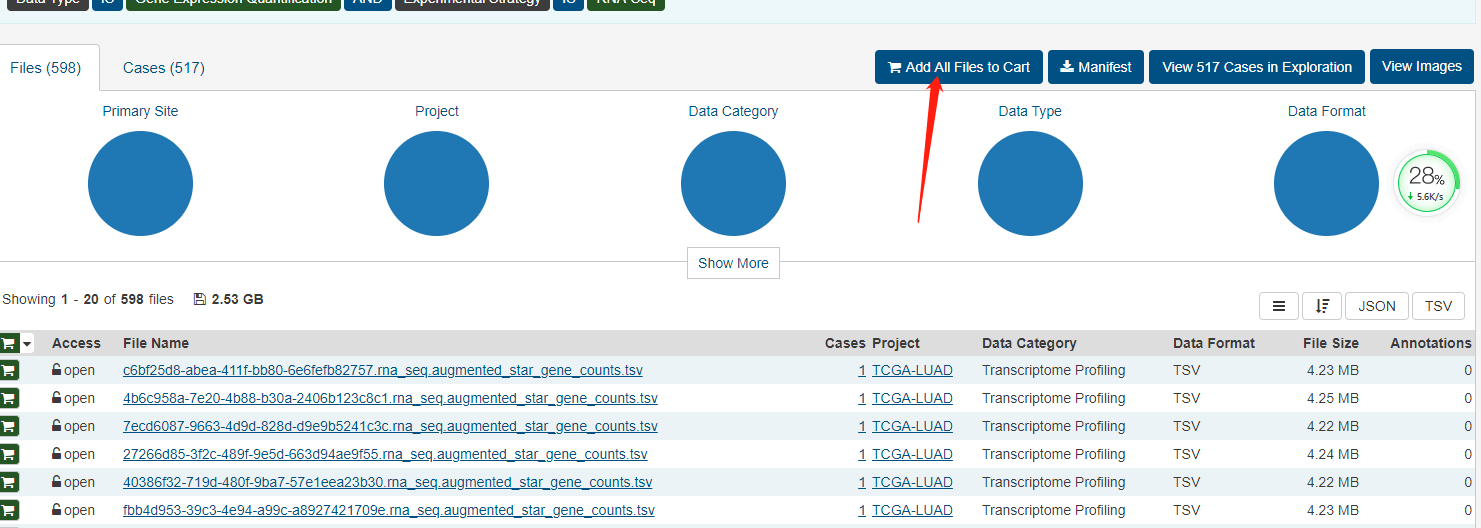
点击顶部Cart,进入下载界面,需要点击这三个地方下载临床数据(Clinical)、json文件(包括文件信息和样本barcode的关系)、表达文件(Download?Cart)。
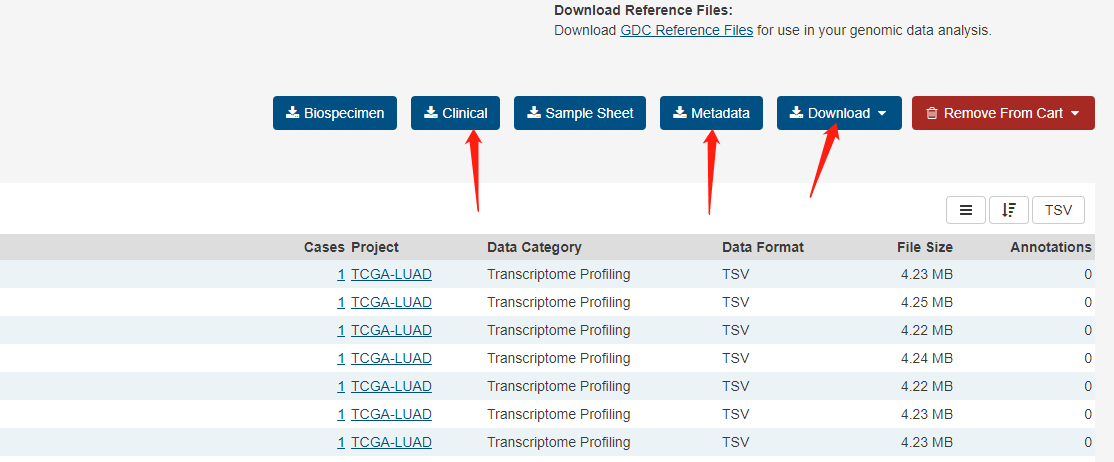
手动解压临床数据文件和json文件,最终我们得到以下三个文件:
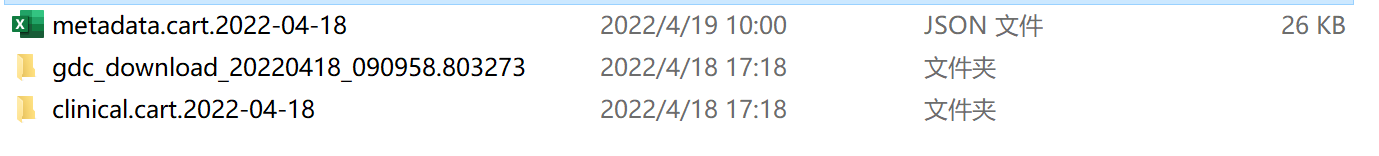
到此为止我们下载好了所需数据然后进行数据整理,
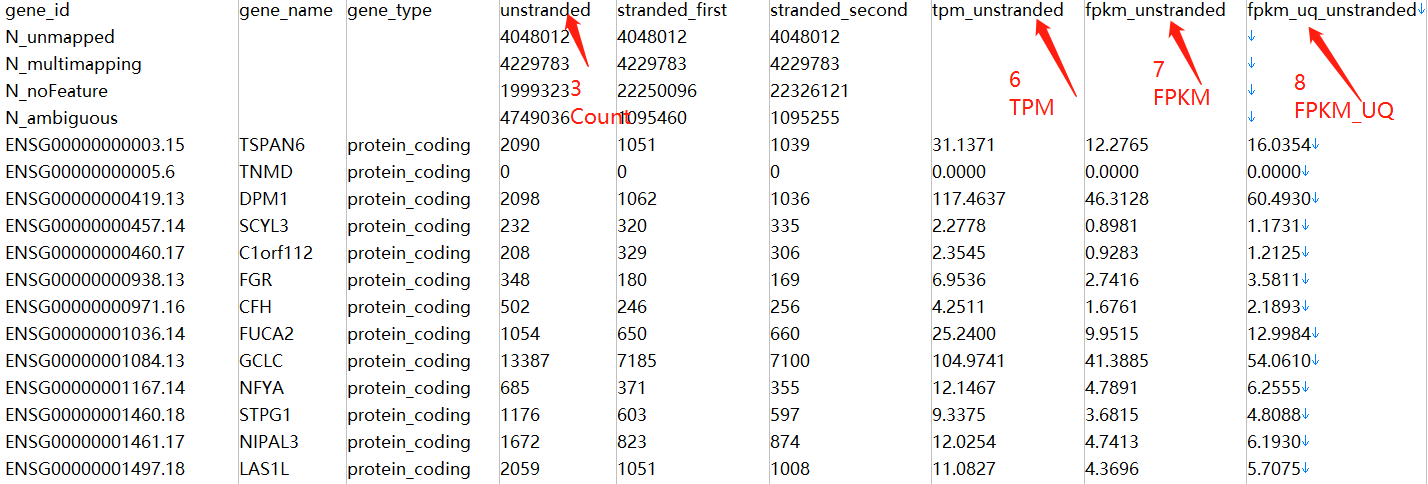
Tips: 此处不需要将下载的tsv文件合并到一个文件夹中,如果合并了,会出现样本名称全部为NA
如果已合并,需要对应修改count_file_name <- sapply(count_file_name,function(x){x[2]})为count_file_name <- sapply(count_file_name,function(x){x[1]})
完整代码:
setwd("你的下载数据路径")
#install.packages("rjson")
library("rjson")
json <- jsonlite::fromJSON("metadata.cart.2022-04-18.json")
View(json)
#id <- json$associated_entities[[1]][,1]
sample_id <- sapply(json$associated_entities,function(x){x[,1]})
file_sample <- data.frame(sample_id,file_name=json$file_name)
#获取gdc_download文件夹下的所有TSV表达文件的 路径+文件名
count_file <- list.files('gdc_download_20220418_090958.803273',pattern = '*.tsv',recursive = TRUE)
#在count_file中分割出文件名
count_file_name <- strsplit(count_file,split='/')
count_file_name <- sapply(count_file_name,function(x){x[2]})
matrix = data.frame(matrix(nrow=60660,ncol=0))
for (i in 1:length(count_file)){
path = paste0('gdc_download_20220418_090958.803273//',count_file[i])
data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
colnames(data)<-data[2,]
data <-data[-c(1:6),]
data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
matrix <- cbind(matrix,data)
}
write.csv(matrix,'COUNT_matrix.csv',row.names = TRUE)
设置Gene Symbol为列名
#------------------------------增加部分:设置Gene Symbol为列名的矩阵(前面得到的是Ensembl ID)------------------------------------------
path = paste0('gdc_download_20220418_090958.803273//',count_file[1])
data<- as.matrix(read.delim(path,fill = TRUE,header = FALSE,row.names = 1))
gene_name <-data[-c(1:6),1]
matrix0 <- cbind(gene_name,matrix)
#将gene_name列去除重复的基因,保留每个基因最大表达量结果
matrix0 <- aggregate( . ~ gene_name,data=matrix0, max)
#将gene_name列设为行名
rownames(matrix0) <- matrix0[,1]
matrix0 <- matrix0[,-1]
分为normal和tumor矩阵
#------------------------------增加部分:分为normal和tumor矩阵--------------------------
sample <- colnames(matrix0)
normal <- c()
tumor <- c()
for (i in 1:length(sample)){
if((substring(colnames(matrix0)[i],14,15)>10)){ #14、15位置大于10的为normal样本
normal <- append(normal,sample[i])
} else {
tumor <- append(tumor,sample[i])
}
}
tumor_matrix <- matrix0[,tumor]
normal_matrix <- matrix0[,normal]
#写入文件
临床数据整合
setwd("你的路径")
#install.packages("rjson")
library("rjson")
json <- jsonlite::fromJSON("metadata.cart.2022-04-18.json")
View(json)
entity_submitter_id <- sapply(json$associated_entities,function(x){x[,1]})
case_id <- sapply(json$associated_entities,function(x){x[,3]})
sample_case <- t(rbind(entity_submitter_id,case_id))
clinical <- read.delim('clinical.cart.2022-04-18\\clinical.tsv',header = T)
clinical <- as.data.frame(clinical[duplicated(clinical$case_id),])
clinical_matrix <- merge(sample_case,clinical,by="case_id",all.x=T)
clinical_matrix <- clinical_matrix[,-1]miRNA数据整合
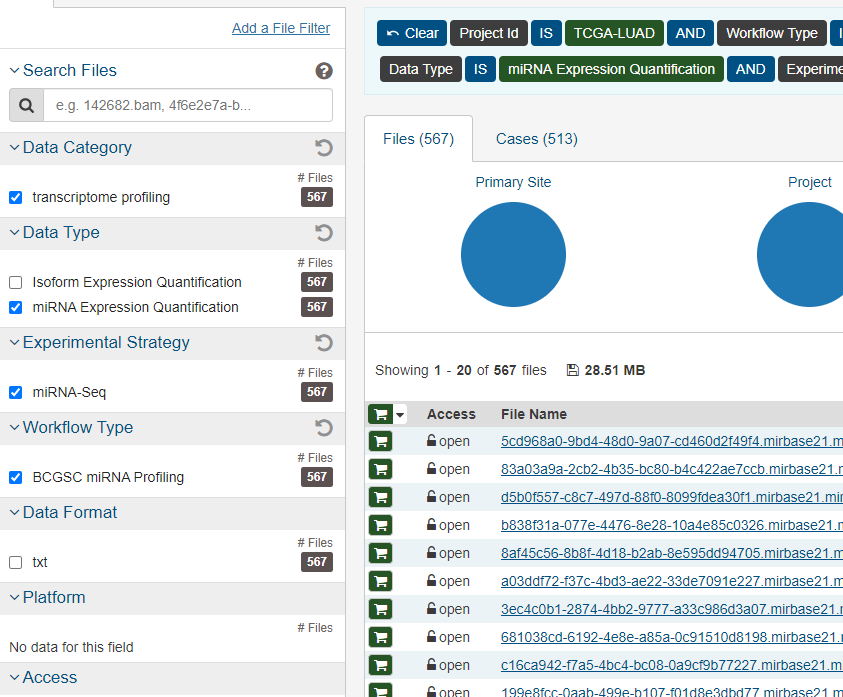
library("rjson")
json <- jsonlite::fromJSON("metadata.cart.2022-09-27.json")
View(json)
#id <- json$associated_entities[[1]][,1]
sample_id <- sapply(json$associated_entities,function(x){x[,1]})
file_sample <- data.frame(sample_id,file_name=json$file_name)
#获取gdc_download文件夹下的所有miRNA表达文件的 路径+文件名
count_file <- list.files('gdc_download_20220927_150057.906231',pattern = '*quantification.txt',recursive = TRUE)
#在count_file中分割出文件名
count_file_name <- strsplit(count_file,split='/')
count_file_name <- sapply(count_file_name,function(x){x[2]})
matrix = data.frame(matrix(nrow=1881,ncol=0))
for (i in 1:length(count_file)){
path = paste0('gdc_download_20220927_150057.906231//',count_file[i])
data<- read.delim(path,fill = TRUE,header = T,row.names = 1)
data <- data[1] #取出count列(第1列),rpm列(第2列)
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
matrix <- cbind(matrix,data)
}
王老师您好,非常抱歉打扰您,我在安装TCGAbiolinks包时,TCGAbiolinksGUI.data在安装时总是显示退出时值不为0,一般什么情况导致的,一般怎么解决?
R已经升级到最新版本了。而且在biocoductor上下载的TCGAbiolinksGUI.data.tar 在Rstudio里 也装不上,同样显示退出时值不是0。该怎么办?非常感谢
额 这可能是版本的原因,R语言安装包经常会遇到这个问题 或者下载binary包,从binary进行安装,你搜一下 试一下
老师您好,我想知道得到的表达矩阵里边很多基因的表达量是0,我该怎么筛选呢?
你想将他们变成NA不去统计的话:data[data == 0] <- NA
谢谢老师的回复!我是想把矩阵里边的表达量多数是0的基因去掉,但是我也不好确定是表达量0占比多少才删,如果是人为确定条件的话,我想把每一列基因对应的表达值只要是0的占比超过30%就去掉?可能得需要在前边的完整代码里补充一下
进哥,请问你给出的代码中出现这个报错怎么解决
Error in data[-c(1:6), 1] : object of type ‘closure’ is not subsettable
你好,哪一步的?data[-c(1:6),]?不应该会有问题,还没解决的话加微信看看
进哥,如何加微信询问问题呢?
首页我的简历有电话,我直接发你也行18021308280
老师您好,请问我倒数第二步一直报错,是为什么呢
In file(file, “rt”) :
cannot open file ‘gdc_download_20220418_090958.803273//0052ae83-7ae5-470a-a125-5cd94a9fa9e9/a6a6b9c6-9db7-42b3-a09f-770b7e126fbb.rna_seq.augmented_star_gene_counts.tsv’: No such file or directory
解决了吗 路径设计有问题,还有问题加微信
谢谢您 已经解决了,有个其他的问题想要请教您,最近想找肿瘤耐药的临床样本 感觉这个很少,请问您有什么数据库推荐的吗?
我也遇到了这个问题,请问是怎么解决的。谢谢!
请问问题解决了吗?我也遇到了这个问题
什么问题 还没没解决的话可以进群讨论
https://www.jingege.wang/jingle_science/
进哥,运行到4行的时候显示报了这个错误,请问是不是我的这个json文件有问题啊?跟电脑文件储存的路径会有关系吗?
> json <- jsonlite::fromJSON("metadata.cart.2023-03-28.json")
Error: lexical error: invalid char in json text.
metadata.cart.2023-03-28.json
(right here) ——^
搞定了吗?没搞定加微信发我文件看看
搞定了已经,还有一个问题进哥,gene symbol设为列名之后输出的文件是csv文件,我想接着往后运行差异分析,但是我看别的教程的up主他们的数据矩阵都是txt文件,这个后面应该怎么接着运行呢?
文件读取换成read.csv读入就可以
咋解决的呀
你好,我想请问下,gdc上下载的临床信息没有生存结局的数据(OS,DSS,DFI和PFI)等,那是去哪里下载,xena上的好像不是最新的。现在的情况是我用R包下载的临床数据却和xena上另外下载的生存信息匹配后有NA值,说明不是同个版本的?需要怎么找到最新的生存结局数据呢?
你好 如果需要最新的 可能需要自己计算了,参考数据原文:Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V; Cancer Genome Atlas Research Network, Hu H. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018 Apr 5;173(2):400-416.e11. doi: 10.1016/j.cell.2018.02.052
老师 我想问一下 做cerna 下载rna-seq中可以提取mirna的表达量,那还用单独下载mirna的表达量吗?
当然,使用miRNA测序数据,转录组中应该只有部分 不全的
王老师,想问修改了相同counts的文件名后,报错了
Error in type.convert.default(data[[i]], as.is = as.is[i], dec = dec, :
invalid multibyte string at ‘ci’
In addition: Warning messages:
1: In read.table(file = file, header = header, sep = sep, quote = quote, :
line 3 appears to contain embedded nulls
下一步该怎么解决
Error in type.convert.default(data[[i]], as.is = as.is[i], dec = dec, :
invalid multibyte string at ‘ci’
In addition: Warning messages:
1: In read.table(file = file, header = header, sep = sep, quote = quote, :
line 3 appears to contain embedded nulls
2: In read.table(file = file, header = header, sep = sep, quote = quote, :
line 5 appears to contain embedded nulls
3: In scan(file = file, what = what, sep = sep, quote = quote, dec = dec, :
embedded nul(s) found in input
倒数第二步咋这样啦,
看不出来问题,要不加我微信,我明天有空远程看看?
王老师,我有两个问题想请教
1、我是用TCGAbiolinks包下载的数据,但是中途有部分文件下载失败,请问要怎么查找下载失败的文件信息然后补充下载?
2、我在网站上下载json文件时,网站提示“GDC download service is currently experiencing issues”,请问这是我的问题还是网站的问题呢?
王老师,第一个问题已经解决,我发现只要文件储存地址设置一致,R包会只补充下载漏掉的文件
好的 解决就好
第二个问题看提示应该是网站问题 你换个浏览器或者网络看看 不行就过一段时间再试试
好的,谢谢老师,json文件除了官网还有别的什么途径下载吗
应该没有吧,这其实是一个样本名文件名对应的文件,需要和你的下载对应
王老师,想请问一下,数据下载好了,但是在提取单基因差异表达分析后,出现null device 1,图片是ns,试了好几个基因结果都是一样,不知道是否是代码出了问题,能否出个教程?感谢!
请问解决了吗 前两天忙 刚刚看见,代码没有问题 绘图保存出了问题 确认你的图片保存路径是否正确?如果还有问题 加我微信我看看
我也遇到了相同的问题。。。。
什么问题 解决了吗 没有的话加微信讨论
王老师,想请教下如何在TCGA中下载结直肠癌的数据,是分别下载结肠癌的和直肠癌的数据,然后再进行合并吗。谢谢。
是的 可以COAD 和READ分别下载进行合并 或者直接两种全部加入到cart 按照同样方式提取合并即可
欧欧,那问下王老师,直接两种全部加入到 cart中,是不是直接点TCGA首页中Case by Major Primary Site 中的colorctal cancer,还是说需要通过输入TCGA-COAD AND TCGA-READ呢,谢谢
你先试一下 按照单个检索之后分别加入cart 然后进入cart一起下载
再有问题直接微信吧
好的,谢谢老师
进哥好,在第一个MATRIX运行后出现Error in file(file, “rt”) : cannot open the connection
In addition: Warning message:
In file(file, “rt”) :
cannot open file ‘gdc_download_20221030_023742.984354//NA’: No such file or directory
怎么处理?麻烦进哥了
你好,查看一下你的count_file_name 中的文件名是否存在非数据文件的,因为你这里出现了gdc_download_20221030_023742.984354//NA,这个NA,搞不定加微信
微信怎么加?
网站顶上我的简历里面有微信二维码 我的简历也有手机号
请问你解决了吗?
你也是一样的问题吗?需要的话加我微信我看看
说错了,是没有列名,只有行(基因)名和COUNT数
老师您好,我按照您的方法进行操作之后第一个matrix命令得到的矩阵没有行的名字,列的名字存在,这是什么问题啊?
你是不是把下载的tsv文件放到一个文件夹下了,如果是就会出现这样的问题,需要修改代码,把file_name<-....最后[2]变成[1]
如果自己搞不定加我微信,给你看看
请问这个报错什么意思,Error in order(list(“bce25281-502e-4599-9679-32dc8462ffb1”, “7fb03840-5153-4dc6-a302-5a89aa4e1fb6”, :
unimplemented type ‘list’ in ‘orderVector1’
目前具体我也看不出来,方便的话加我微信看看
您好请问这是为什么?一直说我Error in read.table(file = file, header = header, sep = sep, quote = quote, :
‘row.names’里不能有重复的名字
setwd(“C:\Users\samsung\Desktop\rawdate1”)
#install.packages(“rjson”)
library(“rjson”)
json <- jsonlite::fromJSON("metadata.cart.2022-10-20.json")
View(json)
#id <- json$associated_entities[[1]][,1]
sample_id <- sapply(json$associated_entities,function(x){x[,1]})
file_sample <- data.frame(sample_id,file_name=json$file_name)
#获取gdc_download文件夹下的所有TSV表达文件的 路径+文件名
count_file <- list.files('rawdata',pattern = '*.txt',recursive = TRUE)
#在count_file中分割出文件名
count_file_name <- strsplit(count_file,split='/')
count_file_name <- sapply(count_file_name,function(x){x[2]})
matrix = data.frame(matrix(nrow=1714,ncol=0))
for (i in 1:length(count_file)){
path = paste0('rawdata//',count_file[i])
data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
colnames(data)<-data[2,]
data <-data[-c(1:6),]
data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
matrix <- cbind(matrix,data)
}
write.csv(matrix,'COUNT_matrix.csv',row.names = TRUE)
你好 你下载的什么数据?提示行名重复,也就是基因名称重复 如果确实如此 需要换一下数据读取策略
R version 4.2.1 (2022-06-23 ucrt) — “Funny-Looking Kid”
Copyright (C) 2022 The R Foundation for Statistical Computing
Platform: x86_64-w64-mingw32/x64 (64-bit)
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under certain conditions.
Type ‘license()’ or ‘licence()’ for distribution details.
R is a collaborative project with many contributors.
Type ‘contributors()’ for more information and
‘citation()’ on how to cite R or R packages in publications.
Type ‘demo()’ for some demos, ‘help()’ for on-line help, or
‘help.start()’ for an HTML browser interface to help.
Type ‘q()’ to quit R.
[Workspace loaded from ~/.RData]
> setwd(“D:\\BRCA”)
> #install.packages(“rjson”)
> library(“rjson”)
> json View(json)
> #id sample_id file_sample #获取gdc_download文件夹下的所有TSV表达文件的 路径+文件名
> count_file #在count_file中分割出文件名
> count_file_name count_file_name matrix=data.frame(matrix(nrow=60660,ncol=0))
> for (i in 1:length(count_file)){
+ path = paste0(‘gdc_download_20221006_102846.585898//’,count_file[i])
+ data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
+ colnames(data)<-data[2,]
+ data <-data[-c(1:6),]
+ data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
+ colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
+ matrix View(matrix)
这个报错是什么意思啊
你好,需要看具体报错信息的,这样看不出来问题
> setwd(“D:\\BRCA”)
> #install.packages(“rjson”)
> library(“rjson”)
> json View(json)
> #id sample_id file_sample #获取gdc_download文件夹下的所有TSV表达文件的 路径+文件名
> count_file #在count_file中分割出文件名
> count_file_name count_file_name matrix=data.frame(matrix(nrow=60660,ncol=0))
> for (i in 1:length(count_file)){
+ path = paste0(‘gdc_download_20221006_102846.585898//’,count_file[i])
+ data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
+ colnames(data)<-data[2,]
+ data <-data[-c(1:6),]
+ data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
+ colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
+ matrix View(matrix)
我的意思是报错信息 就是运行代码之后的报错
搞不定的话加我微信
Error in data.frame(…, check.names = FALSE) :
参数值意味着不同的行数: 60660, 51065
这样的话你需要把上面代码里的60660改成51065,你确认一下每个数据文件里行数是多少(R语言或者Excel打开查看) 这个60660根据你的实际行数进行修改
我不清楚你是不是下载的转录组测序数据,这个应该是60660,如果其它数据,按照我上面所说的修改60660成实际行数
老师您好,我也遇到了这个报错 我下载的是转录组数据,他报错:
Error in data.frame(…, check.names = FALSE) :
参数值意味着不同的行数: 60660, 29397
于是我改成了29397;运行后又报错:
data.frame(…, check.names = FALSE) :
参数值意味着不同的行数:29397, 60660。
请问这到底怎么回事呢?
而且最后处理只有50个样本数,但是实际上是有1000+样本的,请问还有哪里需要改代码吗?运行的是您给的代码
您好 得根据你的数据进行代码修改 加我微信吧 我远程给你看看
老师你好,在做临床数据整合的时候最后两步报错
Error in sort.list(bx[m$xi]) :
‘x’ must be atomic for ‘sort.list’, method “shell” and “quick”
Have you called ‘sort’ on a list?
请问怎么解决呢?老师的代码给了莫大帮助,谢谢老师!
你好,这个临床数据整合代码有点多余 其实用Excel就可以很快实现,可以用Excel打开tsv文件的,所有信息都在这个文件里。具体出错原因需要确定的话加我微信我看看你的数据
还有就是nrow=60660,这个数字是随便写的还是咋确定呢?
这个数值是表达数据文件中的所有基因数目,转录组是60660,其它数据需要打开下载的任意一个文件看一下有几行
你好进哥,设置列名的那个增加代码是单独运行还是要整合到完整代码里面使用呢?
你好,这个在吗是加在上面代码for循环中的,你先理解一下,不懂再问
进哥你好,我用这个临床代码提取后发现矩阵文件缺少了分级数据,然后看了一下clinical里面的内容发现没有grade项目,我该怎么解决
这种肿瘤是应该有grade数据是吧?没有的话就是没有了,还是你指的是stage
请问这要怎么办
> clinical <- read.delim("clinical.cart.2022-09-23\\clinical.tsv",header = TRUE)
Error in file(file, "rt") : cannot open the connection
In addition: Warning message:
In file(file, "rt") :
cannot open file 'clinical.cart.2022-09-23\clinical.tsv': No such file or directory
这个提示就是找不到你的文件,确认你的路径和文件名是否正确
现在又报另一个问题了Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec, :
line 1 did not have 159 elements
进哥,您哈,看了你的代码下载普通的临床信息没问题,但是想从官网下载整理用药信息您可以分享下方法吗 ?
您好,这个没有注意过 不过我打开下载的tsv格式临床数据文件,在145列往后就是相关用药史和放化疗信息,不过我下载的几种肿瘤都是这些信息缺失,您看一下,不对我再看看
> matrix = data.frame(matrix(nrow=60660,ncol=0))
> for (i in 1:length(count_file)){
+ path = paste0(‘gdc_download_20220918_132616.576012’,count_file[i])
+ data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
+ colnames(data)<-data[2,]
+ data <-data[-c(1:6),]
+ data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
+ colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
+ matrix <- cbind(matrix,data)
+ }
Error in file(file, "rt") : cannot open the connection
In addition: Warning message:
In file(file, "rt") :
cannot open file 'gdc_download_20220918_132616.5760120022cd20-f64f-4773-b9ff-a3de0b71b259/8d1641ea-7552-4d23-9298-094e0056386a.rna_seq.augmented_star_gene_counts.tsv': No such file or directory
您好,请问无法读取文件怎么解决呢?
路径和文件夹都是对的,把文件夹删了另一个文件夹也是同样的报错。matrix文件里是空的
我也是,请问下您是怎么解决的?
你好,这个原因多种,反正就是对应目录下没有数据,核对一下路径有没有问题 不清楚加我微信,我的简历里面有
你好 加我微信吧 上午给你看看
老师,请问一下。matrix 60660 obs. Of 0 variables。为什么我的是0呀,而且matrix打开后是空白?
你好 就这样我看不出问题 方便的话加我微信 给你看看
请问临床数据的case id是啥
临床数据整合这个代码很多余 Excel其实就可以打开整理
就这个case id 是指的每个患者的ID,TCGA-***-***-这个是样本编号,一个患者可能有癌旁组织和癌组织,而临床信息是一个患者对应一条 差别在这儿
老师我换了这的代码跟之前一样只出来了一列数据,for后面改了1:150,metadata也跟评论里有一样问题的同学一样重新下过了,还是只有一列数据
诶 这样说我看不出问题 要不加我微信
博主您好,请问我运行到第四行的时候,提示这样的错误是什么意思呢?
> json <- jsonlite::fromJSON("metadata.cart.2022-09-14.json")
Error in loadNamespace(x) : 不存在叫‘jsonlite’这个名字的程辑包
您好 需要安装一下这个包:install.packages(“jsonlite”)
可以將
count_file_name <- sapply(count_file_name,function(x){x[2]})
的 function(x){x[2]} 改為 function(x){x[1]} 試試
目的是什么?2是文件名,1是文件夹名
上面有朋友提到會顯示NA,這樣改改看即可
老师能不能出个从xena数据库下载的TCGA数据来分析差异基因与临床特征之间关系的教程呢?谢谢老师了!
这个分析比较简单,不需要R,excel就可以实现匹配和差异分析,你现在问题在哪一步
就是目的基因表达高低与肿瘤患者年龄、性别、分期等临床特征之间的关系,相当于一个基线资料表?我好像表达的不太清楚(哭/(ㄒoㄒ)/~~
https://www.zhihu.com/question/519669640
就是这个知乎问题里面的这种分析要怎么做呢?求老师指导,谢谢~
这个分析不需要R 没有必要 方便的话加我微信交流
老师你好,按上述代码运行后生成的表格只有一列序号是为什么呢
解决啦,重新下载了一下metadata文件就好了,谢谢老师
不客气
你好,请问怎么样能把mRNA的数据单独提取出来呢?
第三列genetype,提取数据的时候加上data <- data[which(data$gene_type == "protein_coding"),] 即 data <- data[which(data$gene_type == "protein_coding"),] data <-data[-c(1:6),] data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
感谢!
我发现输入了这个data <- data[which(data$gene_type == "protein_coding"),]之后,好像就不需要 data <-data[-c(1:6),] 这一步了,前面6行也一并被筛选掉了~
这样要报错,说是参数值意味着不同的行数。。。
有可能你下载的数据里面有不是表达量的文件,方便的话加我微信讨论
下载的全是转录组表达量的数据,里面有mRNA和IncRNA的,用你提供的那一行代码筛选后mRNA数量是19962行,所以我试着把matrix = data.frame(matrix(nrow=60660,ncol=0))中的60660改成19962,就行了,而且data <-data[-c(1:6),]这一行也要删了,不然结果会少6行,进哥你看这样是否正确呢
matrix = data.frame(matrix(nrow=60660,ncol=0))
for (i in 1:length(count_file)){
path = paste0(‘gdc_download_20220418_090958.803273//’,count_file[i])
data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
colnames(data)<-data[2,]
data <-data[-c(1:6),]
data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
matrix <- cbind(matrix,data)
}
您好,请问想提取fpkm数据,将for循环的第3列改为第7列后还是不能提取,还需要改动哪里呢?麻烦您了谢谢
不用的,你逐步运行到data <-data[-c(1:6),] 看一下data数据里面,确认一下数据以及FPKM第几列,不明白加我微信
我临床矩阵出来的数据是NA
你先确认metadata文件和clinic文件都是一种癌症类型,搞不定加微信讨论 另外多说一句 这个临床数据整合代码有点多余
我也遇到同样问题,count_file_name执行出来是NA。是什么原因呢?
那就是count_file没有读取到你的目标目录下的文件,先确认一下你的目录和文件夹名称,没问题的话加微信讨论
谢谢,是这个问题。已经解决
您好,感谢您的分享!希望可以继续分享整理肿瘤突变数据的代码?
谢谢,好的 后续有时间整理
clinical <- as.data.frame(clinical[duplicated(clinical$case_id),]) #是不是应该为!duplicated() 来去除重复呢
准确来说是的应该为!duplicated() 实际上这个clinical整合的代码 我觉得很多余
是的,用于数据分析后面我还是提取了生存时间、状态以及分期分级的信息。BTW,我看到好像很多肿瘤的Grade信息不再提供了。至少乳腺癌和肾细胞癌是这样的。
你好博主 在临床信息整理中。clinical <- as.data.frame(clinical[duplicated(clinical$case_id),]) 其中duplicate() 去重复是否应该为!duplicate()呢,只使用duplicate()是否是取了重复的值呢?
您好,下载的时候clinic信息都duplicate了,所以duplicate()和!duplicated() 的结果应该一样,准确说的话应该是!duplicated()
老师您好,我在合并时出现显示这种错误,老师您可以帮忙解答一下吗
clinical_matrix <- merge(sample_case,clinical,by="case_id",all.x=T)
Error in sort.list(bx[m$xi]) :
'x' must be atomic for 'sort.list', method "shell" and "quick"
Have you called 'sort' on a list?
您好 这样我也不清楚,还没解决的话要不你加我微信,发我文件看一下
进哥哥,我是通过GDCquery分别下载了肿瘤和正常的表达矩阵,但是我发现两者直接基因数量有差异,肿瘤样本里可能第一行基因名有4万,而正常样本里只有3万多了,这是合理的吗?
您好,这个没有关系,你下载的第一列基因名应该是ensembl transcript ID吧,反正按照这一列合并就好。
王老师,您好,麻烦咨询下您,这个TCGA表达矩阵最后处理出来的是原始的count数据吗?还是fpkm?
您好 都有,在一个文件里,您修改提取的列可以得到不同的数据,文中有介绍
请教进哥哥,为什么形成的矩阵没有列名呢?
你好 代码修改过吗?
你把代码发我看看
没有,
setwd(“F:\\Bioresearch”)
#install.packages(“rjson”)
library(“rjson”)
json <- jsonlite::fromJSON("metadata.cart.2022-08-12.json")
View(json)
#id <- json$associated_entities[[1]][,1]
sample_id <- sapply(json$associated_entities,function(x){x[,1]})
file_sample <- data.frame(sample_id,file_name=json$file_name)
#View(file_sample)
#View(sample_id)
#在count_file中分割出文件名
count_file <- list.files('F:\\Bioresearch\\files',pattern = '*.tsv',recursive = TRUE)
count_file_name <- strsplit(count_file,split='/')
count_file_name <- sapply(count_file_name,function(x){x[2]})
count_file_name执行出来是NA
您方便的话直接加我微信 我远程看一下 18021308280
你好 临床数据整合部分运行到clinical <- read.csv("clinical.cart.2022-08-10\\clinical.tsv",header = T)
提示Error in file(file, "rt") : 无法打开链结
此外: Warning message:
In file(file, "rt") :
无法打开文件'clinical.cart.2022-08-10\clinical.tsv': No such file or directory
请问您这是什么原因呢
你好,检查一下对应目录下有没有clinical.tsv文件,是不是没有解压还是路径设置不对?不确定就改成绝对路径
你好 我在TCGA下载我想要的样本后,通过你的代码整理成矩阵 然后还是通过你的代码绘制火山图啥的 但是到 # 差异分析检验 et et <- exactTest(y)
Error in exactTest(y) :
At least one element of given pair is not a group.
Groups are: 0
这个需要怎么解决呢
应该是前面定义tumor normal没有成功,如果可以的话你自己看一下定义分组之后的结果,不行的话加我微信讨论18021308280
你好 我用你的代码定义了分组 发现分组normal 里面没有 全在tumor组里面
那就是没有正常样本,可以结合gtex
你好,我想问一下,for (i in 1:10){
path = paste0(‘gdc_download_20220418_090958.803273//’,count_file[i])
data<- read.delim(path,fill = TRUE,header = FALSE,row.names = 1)
colnames(data)<-data[2,]
data <-data[-c(1:6),]
data <- data[3] #取出unstranded列(第3列),即count数据,对应其它数据
colnames(data) <- file_sample$sample_id[which(file_sample$file_name==count_file_name[i])]
matrix <- cbind(matrix,data)
}这一步之后,matrix没有列名是怎么回事呀?可以解答一下嘛?谢谢!
您好 不好意思 刚刚看到你的问题
你已经把代码中对应文件路径改成你自己的吗?如果解决不了可以加我微信讨论
你好,我也遇到同样的问题,想问一下你的联系方式可以吗
可以,网站上我的简历下面有微信和电话
路径是改为自己的,现在已经用其他的方法解决这个问题了,谢谢您
感觉很详细但是过程中遇到了一些问题,进哥能不能帮忙解答一下呢?
count_file count_file
NULL
上面是一步一步来的 这一步就变成这样了
你确认一下你的路径是否正确,然后这一步的结果count_file <- list.files('gdc_download_20220418_090958.803273',pattern = '*.tsv',recursive = TRUE)是什么?如果文件夹下有文件,就不应该null。
json <- jsonlite::fromJSON("metadata.cart.2022-08-01.json")
Error in parse_con(txt, bigint_as_char) :
parse error: object key and value must be separated by a colon (':')
[{ " ""data_format"": ""TSV"", " " ""access"":
(right here) ——^
求大佬指导下,谢谢了
看起来是你的json文件有问题,可以的话加我微信把文件发我 或者直接发我邮箱jin.wang93@outlook.com
进哥哥可以发一下新版突变数据整理的教程吗?
一样的哇,就是第二步选择突变数据,而不是转录组就好,后面的数据下载整合差不多,你先试试,不行再留言