1. 数据下载
新版TCGA表达mRNA/miRNA和临床数据下载及R语言整合代码 – 王进的个人网站
注意:对于miRNA,有两种data type:成熟体isoform expression quantification & 前体stemloop miRNA expression quantification,根据需求选择合适的数据类型。
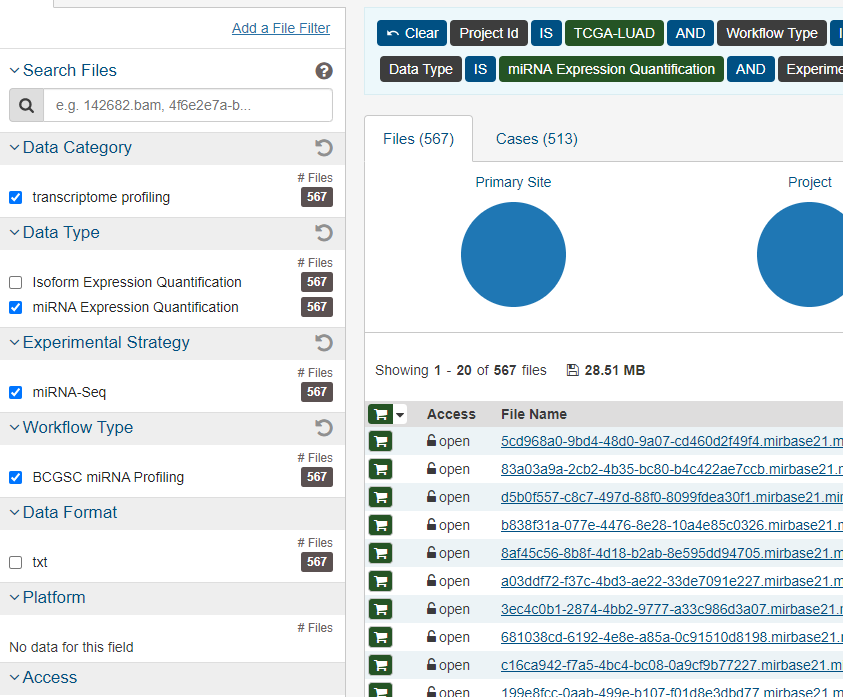
对于表达数据,我们总共需要2个 文件:
json文件(包括文件信息和样本barcode的关系)、表达文件(Download🡪Cart)解压下载的表达文件。
2. 运行APP
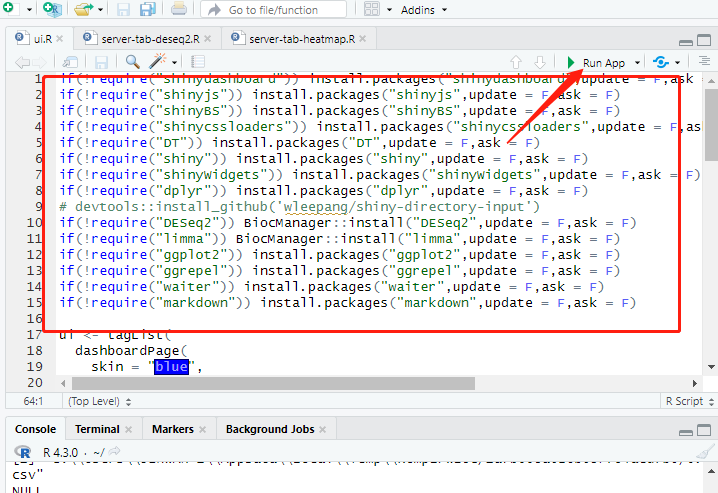
打开shiny APP文件夹中的ui.R文件,我们可以看到需要安装的包,代码窗口右上角有一个run app的按钮,点击该按钮,app自动安装并加载需要的包。
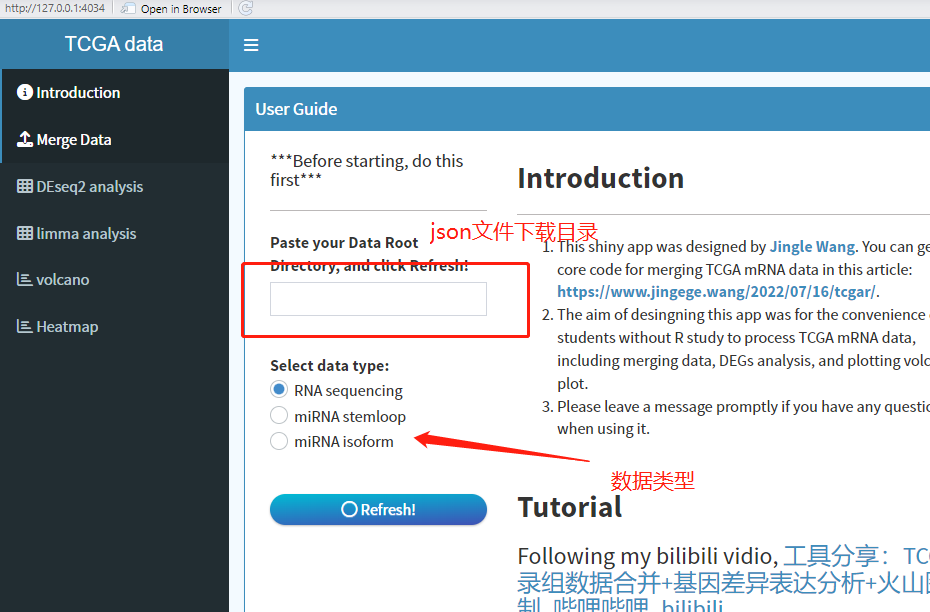
3. 路径及数据类型设置,然后点击Refresh。此处以miRNA isoform 数据为例,选择miRNA isoform。
4. 点击Merge data,app已经自动获取目录下的json文件和数据目录:
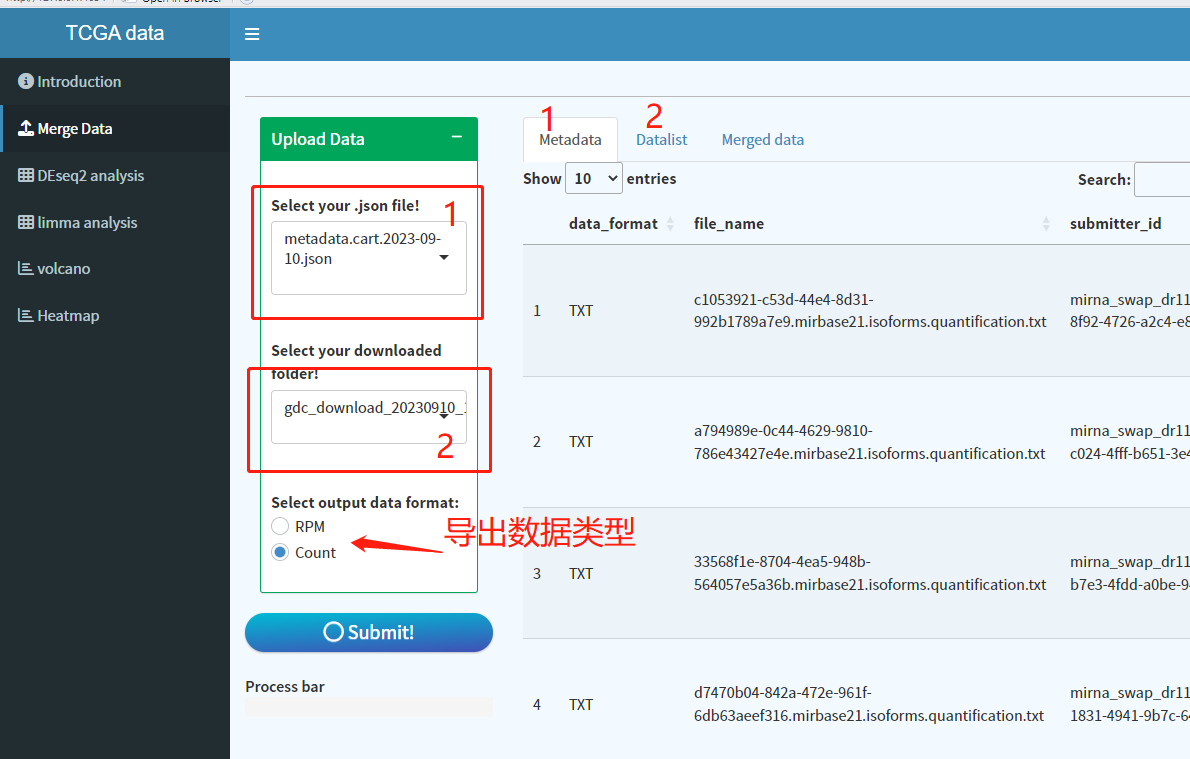
点击Submit,即可自动进行数据合并,合并完成之后,目录下会多出一个整合好的数据文件。
5. DESeq2差异分析
点击DESeq2 analysis,界面非常简洁,选择合并好的count data,点击submit即可。对于刚刚合并的数据,看不到目录下的这个文件,需要回到introduction界面点击refresh按钮即可。
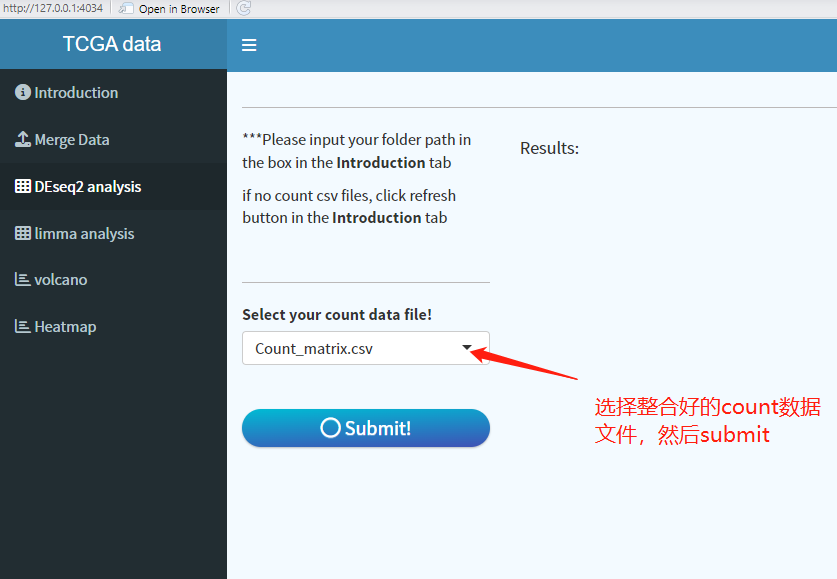
6. 等待片刻,即可得到差异分析的结果。
同时app会自动在设置目录下新建一个result文件夹,并将normalized data和DEG results自动存放进去。注意:DESeq2分析较缓慢,耐心等待结果。
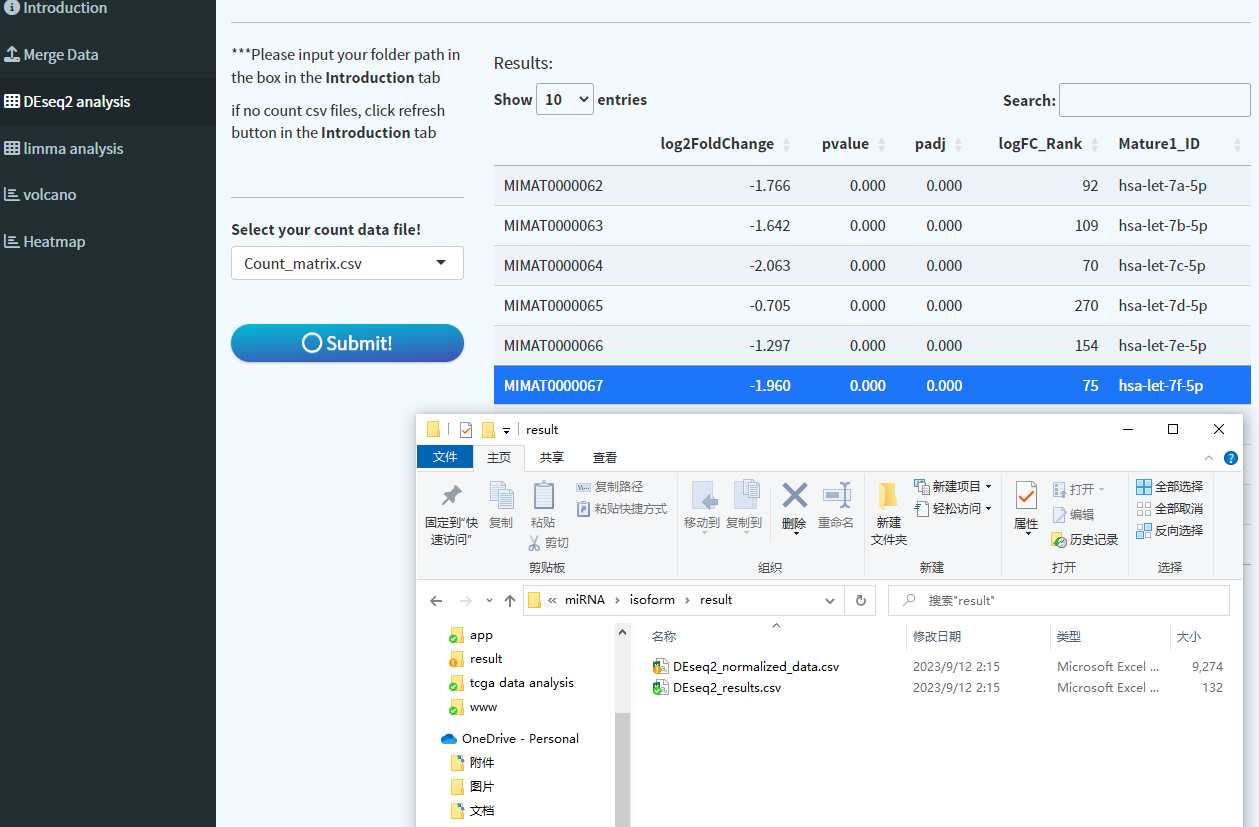
注意:对于mRNA和isoform miRNA,下载的数据中分别以ensembl ID和miRbase ID作为名称,差异分析之后会自动进行ID转换。
对于RNA sequencing结果,ID转化同时会有biotype信息,用于提取lncRNA和mRNA
7. 火山图绘制
点击volcano进入差异基因火山图可视化界面,上传DESeq2差异分析结果,调整相关阈值参数,点击提交即可。
注意:如需对limma包差异分析结果或者自己的差异分析数据进行可视化,需要修改相应列名,使之与DESeq2结果一致。
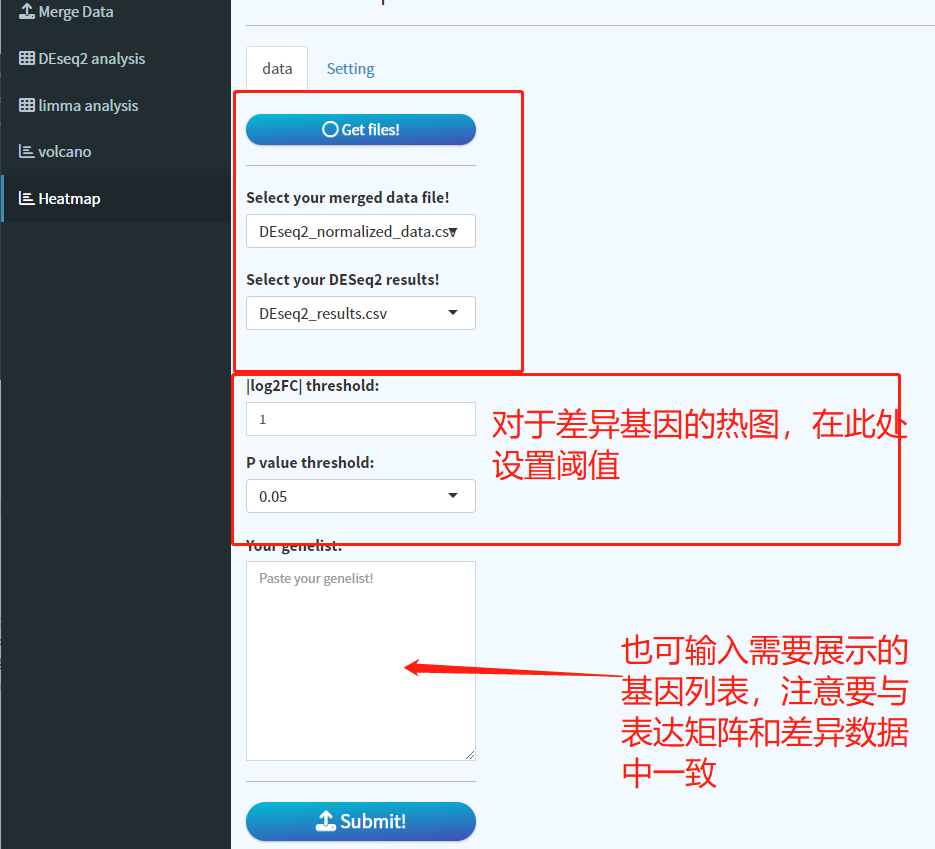
8. 热图绘制
进入Heatmap模块,点击get file,即可自动填充刚刚DESeq2得到的normalized data和DEG文件。点击submit,稍等片刻即可得到热图。Setting中可以进行相关参数的设置。
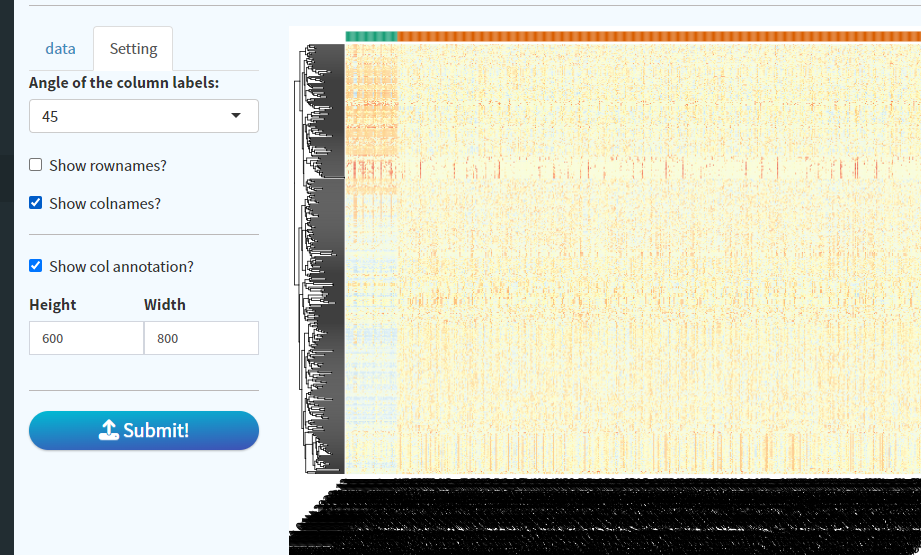
9. 以上分析均以导出count数据为例,大家也可根据自己的需求导出TPM、FPKM或RPM,对于这些数据,需要limma包进行差异分析,APP里面也提供了该模块。如使用limma分析,对于火山图和热图可视化,需要手动修改差异分析结果的列名与deseq2一致。当然我可以让app自动设置,但是应该用的人不多,所以就先不改了。
如需要app代码进行TCGA数据处理,请至:
B站代码获取 – 王进的个人网站 (ID11,已更新至最新三合一的app)
差异分析的表达矩阵里面有重复的基因名字,这样的该如何进行下一步?这个app之后得到的差异表达文件该怎么用r去处理得到一个可以用作绘制火山图的数据呢?
为什么网页版,我点击refresh之后一直没有反应啊?然后R一直闪退?
The sample size in Metadata isn’t equal to the downloaded TSV files, please comfirm!进哥,我用这个小程序,上传metadata .json和downloaded folder后总会出现这个提示什么原因