转载自《生信技能树》
药物预测需要训练集,一般来说推荐使用权威资源作为训练集建好模型,这样就可以去预测你自己的数据。
权威的药物预测训练集资源
那么,比较权威的资源一般就是Cancer Therapeutics Response Portal (CTRP) 和 Genomics of Drug Sensitivity in Cancer (GDSC)
Cancer Therapeutics Response Portal (CTRP)
目前主要是CTRP v2,官网是:http://portals.broadinstitute.org/ctrp.v2.1/
- 481 compounds X 860 CCLs
- correlations to copy-number and gene-expression data
- mutation data integrate CCLE and Sanger/MGH calls
- correlation and enrichment analysis on-the-fly
- box-whisker visualization in addition to enrichment heatmaps
- drill-down to scatter plots and concentration-response curves
- flter by lineage/subtype, growth mode
Genomics of Drug Sensitivity in Cancer (GDSC)
官网是:https://www.cancerrxgene.org/
如果是v2的版本,有809 Cell lines 以及 198 Compounds
如果是看v1版本,987 Cell lines 和 367 Compounds
资源都被整理好了
我们这里直接使用R包oncoPredict整理好的这两个数据库的rdata文件,下载链接是:https://osf.io/c6tfx/ ,
oncoPredict
Contributors: Danielle Maeser Robert Gruener
Date created: 2021-03-26 01:39 PM | Last Updated: 2021-08-15 10:44 PM
下载约700M,重要的文件 如下所示;
171M Aug 14 17:10 CTRP2_Expr (RPKM, log2(x+1) Transformed).rds
177M Apr 3 2021 CTRP2_Expr (TPM, log2(x+1) Transformed).rds
1.1M Apr 3 2021 CTRP2_Res.rds
119M Apr 3 2021 GDSC1_Expr (RMA Normalized and Log Transformed).rds
2.0M Apr 3 2021 GDSC1_Res.rds
100M Apr 3 2021 GDSC2_Expr (RMA Normalized and Log Transformed).rds
906K Apr 3 2021 GDSC2_Res.rds
可以看到 Cancer Therapeutics Response Portal (CTRP) 数据库里面的细胞系表达量矩阵是来自于转录组测序, 所以 提供了 FPKM和TPM两个版本供用户选择。
然后呢 Genomics of Drug Sensitivity in Cancer (GDSC) 数据库里面的细胞系表达量矩阵应该是芯片,因为它使用了 RMA Normalized and Log Transformed ,标准的芯片数据处理方法。
代码探索 (GDSC) 数据库
直接看 v2的版本,有809 Cell lines 以及 198 Compounds
主要是八百多个细胞系的约2万个基因的表达量矩阵,以及对应八百多细胞系的约200个药物的IC50值。
library(reshape2)
library(ggpubr)
th=theme(axis.text.x = element_text(angle = 45,vjust = 0.5))
dir='./DataFiles/Training Data/'
GDSC2_Expr = readRDS(file=file.path(dir,'GDSC2_Expr (RMA Normalized and Log Transformed).rds'))
dim(GDSC2_Expr)
GDSC2_Expr[1:4, 1:4]
boxplot(GDSC2_Expr[,1:4])
df=melt(GDSC2_Expr[,1:4])
head(df)
p1=ggboxplot(df, "Var2", "value") +th
# Read GDSC2 response data. rownames() are samples, colnames() are drugs.
dir
GDSC2_Res = readRDS(file = file.path(dir,"GDSC2_Res.rds"))
dim(GDSC2_Res) # 805 198
GDSC2_Res[1:4, 1:4]
p2=ggboxplot(melt(GDSC2_Res[ , 1:4]), "Var2", "value") +th ; p2
# IMPORTANT note: here I do e^IC50 since the IC50s are actual ln values/log transformed already, and the calcPhenotype function Paul #has will do a power transformation (I assumed it would be better to not have both transformations)
GDSC2_Res <- exp(GDSC2_Res)
p3=ggboxplot(melt(GDSC2_Res[ , 1:4]), "Var2", "value") +th ; p3
library(patchwork)
p1+p2+p3
如下所示 :
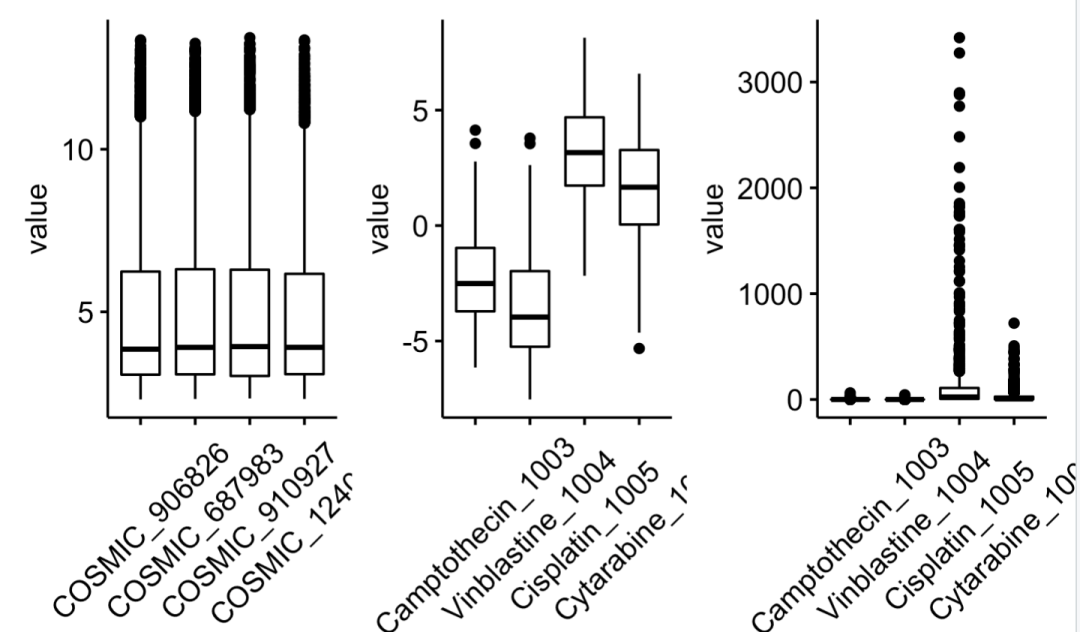
表达量矩阵被归一化很好,就是(RMA Normalized and Log Transformed),跟绝大部分芯片数据分析一样的,介于 0到15之间。
药物的ic50值,最开始的rds文件里面,也就是说从 (GDSC) 数据库下载得到的是被log转换的,所以又重新使用幂函数转回来。
其中半抑制浓度,或称半抑制率,即IC50,其在间接竞争ELISA标准曲线中是一个非常重要的数据。这样就出现了好几个专有名词啦,不过我们可以简单一点理解,就是能杀死癌细胞的药物浓度的一半。
- 比如设计 0.01,0.1,1,10,100 这样的药物梯度
- 发现从浓度1开始可以杀死癌细胞啦,所以我们就认为IC50是 0.5(一般来说不太可能出现浓度更高反而杀不死癌细胞的反常情况)
- 可以看到,浓度 梯度设计决定了 IC50的分辨率,就是一个区间值就足够啦。
- 如果IC50超级高,比如解决好几百大几千说明这是一个废物药物,约等于没有疗效。
- 如果IC50超级低,比如无限接近于0,说明这个药物就是传说中的灵丹妙药!
前面的箱线图,我们展现的是某个药物的八百多细胞系的IC50,这样可以看得出来有一些药物在很多癌症细胞系的表现就是废材,比如 “Cisplatin_1005” 和 “Cytarabine_1006″,当然了,因为我仅仅是展现了4个药物,所以说它们是废材仅仅是相当于 “Camptothecin_1003” 和”Vinblastine_1004″来说。
还有另外一个展现方式,就是看针对具体的细胞系来说,那些药物有奇效那些药物是打酱油。代码如下所示:
ggboxplot(melt(GDSC2_Res[ 1:4 ,]), "Var1", "value") +th 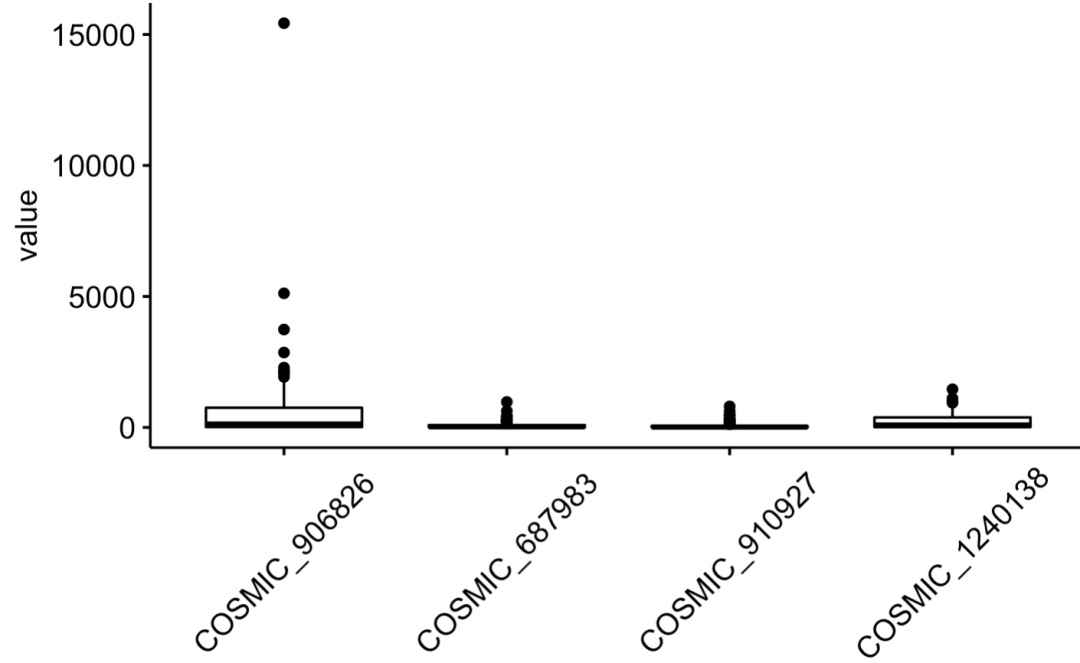
因为每个细胞系的箱线图里面都是约200个药物,所以这样的可视化看不出来具体 的药物表现,并没有太大的意义。我们应该是直接看top药物即可:
round(apply(GDSC2_Res[ 1:4 ,], 1, function(x){
return(c(
head(sort(x)),
tail(sort(x))
))
}),2)
COSMIC_906826 COSMIC_687983 COSMIC_910927 COSMIC_1240138
[1,] 0.00 0.00 0.00 0.05
[2,] 0.00 0.00 0.00 0.07
[3,] 0.01 0.01 0.01 0.09
[4,] 0.04 0.01 0.01 0.21
[5,] 0.05 0.01 0.01 0.95
[6,] 0.05 0.01 0.01 0.98
[7,] 2174.67 310.59 286.42 922.01
[8,] 2285.10 405.44 388.50 925.43
[9,] 2859.17 413.01 471.66 939.02
[10,] 3736.69 436.98 489.76 989.15
[11,] 5118.44 626.42 623.64 1105.89
[12,] 15431.05 973.87 803.89 1457.11
可以看到, 每个细胞系都是有自己的特异性药物和废物药物,IC50接近于0的就是神药,那些大几千的就是辣鸡药物啦。
但是,我们可能是更想看到的是药物名字啦!
apply(GDSC2_Res[ 1:4 ,], 1, function(x){
names(x)=gsub('_[0-9]*','',colnames(GDSC2_Res))
return(c(
names(head(sort(x))),
names(tail(sort(x)))
))
})
COSMIC_906826 COSMIC_687983 COSMIC_910927 COSMIC_1240138
[1,] "Sepantronium bromide" "Daporinad" "Dactinomycin" "Luminespib"
[2,] "Bortezomib" "Sepantronium bromide" "Sepantronium bromide" "CDK9"
[3,] "Dactinomycin" "Dactinomycin" "Bortezomib" "Dinaciclib"
[4,] "Rapamycin" "Bortezomib" "Vinblastine" "Dactinomycin"
[5,] "Dactolisib" "Docetaxel" "Docetaxel" "CDK9"
[6,] "Docetaxel" "Vinblastine" "Luminespib" "Sabutoclax"
[7,] "KU-55933" "EPZ5676" "AZD1208" "IAP"
[8,] "EPZ5676" "Carmustine" "Nelarabine" "Mirin"
[9,] "Acetalax" "Selumetinib" "Doramapimod" "PFI3"
[10,] "Nelarabine" "Nelarabine" "SB216763" "ML323"
[11,] "Doramapimod" "Acetalax" "Carmustine" "AZD5991"
[12,] "Temozolomide" "5-Fluorouracil" "Temozolomide" "Carmustine"
前面的6个药物是各自细胞系的神药,后面的6个是废物药物啦。
现在我们可以尝试一下使用R包之oncoPredict对你的表达量矩阵进行药物反应预测啦!
发表oncoPredict这个包的文献非常新:《oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data》,这个通讯作者就是2014年r包pRRophetic同一个人,相当于是炒冷饭吧!
使用oncoPredict之前先安装,代码如下:
install.packages("oncoPredict") 如果遇到版本问题,请看:https://mp.weixin.qq.com/s/HGfePIQP4yP_nvhjiWdpAQ
使用方法超级简单
首先需要读入训练集的表达量矩阵和药物处理信息,参考前面的教程:药物预测之认识表达量矩阵和药物IC50
rm(list = ls()) ## 魔幻操作,一键清空~
options(stringsAsFactors = F)
library(oncoPredict)
library(data.table)
library(gtools)
library(reshape2)
library(ggpubr)
th=theme(axis.text.x = element_text(angle = 45,vjust = 0.5))
dir='./DataFiles/Training Data/'
GDSC2_Expr = readRDS(file=file.path(dir,'GDSC2_Expr (RMA Normalized and Log Transformed).rds'))
GDSC2_Res = readRDS(file = file.path(dir,"GDSC2_Res.rds"))
GDSC2_Res <- exp(GDSC2_Res)
这里仍然是以Genomics of Drug Sensitivity in Cancer (GDSC) 的v2作为例子,有了训练集的表达量矩阵和药物处理信息,还需要读入你需要做预测的表达量矩阵。
因为我们这个是教程,所以我就不读取自己的表达量矩阵了,直截了当的从Genomics of Drug Sensitivity in Cancer (GDSC) 的v2里面随机挑选10个细胞系作为要预测的矩阵。
testExpr<- GDSC2_Expr[,sample(1:ncol(GDSC2_Expr),10)]
testExpr[1:4,1:4]
colnames(testExpr)=paste0('test',colnames(testExpr))
dim(testExpr)
了训练集的表达量矩阵和药物处理信息,然后也有了待预测的表达量矩阵,接下来就是一个函数的事情啦!这个函数calcPhenotype就是R包 oncoPredict的核心,超级方便!
calcPhenotype(trainingExprData = GDSC2_Expr,
trainingPtype = GDSC2_Res,
testExprData = testExpr,
batchCorrect = 'eb', # "eb" for ComBat
powerTransformPhenotype = TRUE,
removeLowVaryingGenes = 0.2,
minNumSamples = 10,
printOutput = TRUE,
removeLowVaringGenesFrom = 'rawData' )
这个函数运行取决于你的计算资源,需要半个小时左右。好像也没有多线程的可能性,所以只能是慢慢等了,喝一杯咖啡吧,如果可以的话希望你在咱们《生信技能树》公众号任意教程末尾打赏一杯咖啡也行,我们一起慢慢喝,慢慢等!
从函数运行的log日志来看,本质上就是一个岭回归:
17419 gene identifiers overlap between the supplied expression matrices...
Found2batches
Adjusting for0covariate(s) or covariate level(s)
Standardizing Data across genes
Fitting L/S model and finding priors
Finding parametric adjustments
Adjusting the Data
4650 low variabilty genes filtered.
Fitting Ridge Regression model
Calculating predicted phenotype...
Done making prediction for drug 1 of 198
Fitting Ridge Regression model...
Calculating predicted phenotype...
Done making prediction for drug 2 of 198
解读药物预测结果
前面的R包 oncoPredict的核心函数calcPhenotype运行完毕后,会在当前工作目录下面输出 calcPhenotype_Output 文件夹,里面有一个 DrugPredictions.csv的文件,这个都是函数calcPhenotype写死了的。
library(data.table)
testPtype <- fread('./calcPhenotype_Output/DrugPredictions.csv', data.table = F)
testPtype[1:4, 1:4]
补充一下,也可以下载包源码进行修改,包代码是用for循环进行分析的,效率很低,可以进行修改多线程+do.call
另外,不同的数据库资源作为函数的训练集,得到的结果必然是不一样的哦!而且函数也可以调整很多参数。
你好,想请问下一直出现这个报错到底是什么原因啊?,eset已经是matrix格式,换用其他基因集也不行。感谢解答!
result <-calcPhenotype(trainingExprData = GDSC2_Expr,
+ trainingPtype = GDSC2_Res,
+ testExprData = eset,
+ batchCorrect = 'eb', # "eb" for ComBat
+ powerTransformPhenotype = TRUE,
+ removeLowVaryingGenes = 0.2,
+ minNumSamples = 10,
+ printOutput = TRUE,
+ removeLowVaringGenesFrom = 'rawData')
11681 gene identifiers overlap between the supplied expression matrices…
Found2batches
Adjusting for0covariate(s) or covariate level(s)
Standardizing Data across genes
Error in crossprod(design) : "crossprod" is not a BUILTIN function
你好,请确认crossprod函数没有被其它包函数给mask,确认很简单,终端执行crossprod,看这个函数是不是base包的
发现在将 batchCorrect = ‘eb’变成batchCorrect = ‘none’,即可输出结果,请问这样可以吗?
就是不进行去批次,你可以在预测之前进行,eb使用的是combat方法。当然,一个实验的不进行问题不大
同时找寻crossprod是否被mask结果如下
> crossprod
function (x, y = NULL)
.Internal(crossprod(x, y))
请问这个报错怎么处理啊?已确定没有重复命名的数据框了
Found2batches
Adjusting for0covariate(s) or covariate level(s)
Standardizing Data across genes
Error in crossprod(design) : “crossprod” is not a BUILTIN function
确认一下crossprod函数有没有被mask
您好,我用上述代码跑的时候,总是遇到下面的报错:
Calculating predicted phenotype…
Done making prediction for drug 198 of 198
Error in file(file, ifelse(append, “a”, “w”)) :
cannot open the connection
In addition: Warning messages:
1: In dir.create(“./calcPhenotype_Output”) :
cannot create dir ‘./calcPhenotype_Output’, reason ‘Read-only file system’
2: In write.csv(DrugPredictions_mat, file = “./calcPhenotype_Output/DrugPredictions.csv”, :
attempt to set ‘col.names’ ignored
3: In file(file, ifelse(append, “a”, “w”)) :
cannot open file ‘./calcPhenotype_Output/DrugPredictions.csv’: No such file or directory
请问应该怎么解决
进哥,你在视频里不是说score的值越大,敏感性越低吗,到底应该怎么看score的大小呀
进哥,小鼠样本能做这个分析吗,有小鼠的药敏数据库吗
进哥,跟着你的视频学习以后两个数据库都做了,但是还是有问题想请教,用CTRP2的数据做出来的结果该怎么解读:
1. =这个AUC值大小的含义,这个AUC值也是越接近零,说明药物敏感性越强是吗?
2. 结果里的AUC值挺大的,基本都大于1,但是我查了其他文献发现其他文献里的AUC值都在0-1之间,这个是怎么做出来的呀,是把所有结果都映射到0-1这个区间了吗。
谢谢!
进哥,有个问题想请教,药物名称方面的,获得敏感性得分后发现有两个一样的药物,`Oxaliplatin_1089`与
`Oxaliplatin_1806`,`Oxaliplatin_1806`的敏感性得分明显高一些,进哥,这个药物名称后面的数字来源哪里啊,没有找到相关背景知识
进哥,您真是恩人
您好,GDSC中的表达矩阵只有一万到两万个基因,而TCGA下载的表达矩阵以ensembl为geneid,基因数目远超训练集的表达矩阵的基因数目,我需要将TCGA的表达矩阵提取到与GDSC同样的基因数目吗,也就是提取它一万到两万个基因表达的子矩阵?
你好,请问这个问题如何解决:Warning message:
In write.csv(DrugPredictions_mat, file = “./calcPhenotype_Output/DrugPredictions.csv”, :
不能修改’col.names’
貌似不需要理会这个警告信息
王博,请问一下要如何挑选指定癌种的细胞株作为训练组?我在GDSC官网和COSMIC中没有找到COSMIC_906861、COSMIC_1290814等这类名称对应的癌种。
您好,前辈,我不知道为什么我在运行到核心步骤的时候总是报错,在这里卡了好几天了。下面这个报错说的是测试数据集和训练数据集没有办法对应嘛?我应该要怎么解决呢
ERROR: Gene identifiers must be specified as “rownames()” on both training and test expression matrices. Both matices must have the same type of gene identifiers.
CTRP有两个数据格式TPM和RPKM,两个格式的表达量都较大,在使用的时候,是否需要log2或标准化变换呢?
进哥你好,我想问一下,自己的表达矩阵是直接放从GEO下载的一万多个基因,还是放自己最终筛选的基因,但是最终筛选的基因只有8个,使用calcPhenotype函数说,测试集需要至少10个基因,直接放1万多个基因的测试集基因数太多,使用最终的8个基因测试集基因太少,这怎么取舍呢??
哦哦,你没里理解这个包的原理,最好把所有基因放进去分析样本的稳定性,他是根据训练集得到的药物敏感性相关基因的模型进行测试集的计算,得到敏感性之后和你的基因做相关性分析
您好,进哥哥,我得到的药物敏感性结果里面很多NA值,请问这个是为什么啊。
同学,你问题解决嘛
请问这个用tcga表达数据时对输入的格式有要求吗,是fpkm,tpm还是counts,另外tcga数据需要对数化处理吗
你好,要使用标准化之后的矩阵,除了count都可以,log与否影响不大,我是习惯用log 进行正态化
进哥,计算出来有些sencitivity score是负值,这个为什么啊,这样的值应该怎么处理呢?
正常的,这是一个评分,与你的基因比对数据有关,应该是用的log转化的,必然有负值
那负值结果咋个解释啊
进哥,看完您的视频很受启发,单我在输入自己的TCGA TPM格式的数据后,运行calcPhenotype时,总会出现下列问题(基因标识符不匹配的问题),不知道哪里出现了问题,TCGA数据是我处理后的mRNA数据。这是哪里出现问题了呢?往进哥请教
> calcPhenotype(trainingExprData = GDSC2_Expr,
+ trainingPtype = GDSC2_Res,
+ testExprData = testExpr,
+ batchCorrect = ‘eb’, # “eb” for ComBat
+ powerTransformPhenotype = TRUE,
+ removeLowVaryingGenes = 0.2,
+ minNumSamples = 10,
+ printOutput = TRUE,
+ removeLowVaringGenesFrom = ‘rawData’ )
Error in homogenizeData(testExprMat = testExprData, trainExprMat = trainingExprData, :
ERROR: Gene identifiers must be specified as “rownames()” on both training and test expression matrices. Both matices must have the same type of gene identifiers.
Gene identifiers must be specified as “rownames()” on both training and test expression matrices. Both matices must have the same type of gene identifiers.
你测试集的基因名称是Ensembl ID 还是基因名字,现在提示就是基因名称不能对应,还有问题可以进群发图片讨论
https://www.jingege.wang/jingle_science/
我用的就是基因名称,不是ensemble ID
我导入的数据是按下列代码写的,数据准备的和GDSC2_Expr是一致的。因为我总共有484列,就把10改成了484。testExpr1=read.table(“TCGA.txt”,sep=”\t”,header=T,check.names=F)
testExpr1=as.matrix(testExpr1)
testExpr<- testExpr1[,sample(1:ncol(testExpr1),484)]
请问你解决了吗?我也同样的问题卡住了
这不是搬的生信技能树吗?还是注明下出处吧
谢谢提醒,已经补充了
进哥,您给的下载rdata的那个链接打不开,而且这两天不知道为什么GDSC的网站一直进不去训练集下不到,您还有别的获取GDSC数据的方法吗?
OSF | oncoPredict
https://osf.io/c6tfx/可以打开的哇!需要的话我放网盘
可能是我那两天网站维护,这几天又打开了,谢谢进哥
进哥您好,https://osf.io/c6tfx/这个里面的训练集无法下载,提示‘storage.googleapis.com 已拒绝连接。‘
进哥你好,请问在mac上用oncoPredict包的函数calcPhenotype计算药敏时,总是报错error: protect(): protection stack overflow ,有没有不换电脑的解决方案呢,Google了很久都没用
不清楚诶,没有用过,这个方法试过了吗?https://www.coder.work/article/6260809
不行远程我试试 加我微信
请问进哥这个问题解决了嘛,我也遇到这个问题了
哎!硬件配置带不动
可以将calcPhenotype函数的参数设置一下,removeLowVaryingGenes=0.6。https://github.com/maese005/oncoPredict/issues/2,在这个网站里面作者回复了这个问题
香蕉大哥你就是我的神
选定特定的药物后,只有一维数据,Error in trainingPtype[, a] : incorrect number of dimensions,R代码报错,请问老师怎么解决
那你就用两个药物的数据吧,然后提取出来吧
王博你好,请教一下,上述代码的话,如果要换成自己的表达矩阵来预测高低表达组的药物敏感性,只需要把calcPhenotype函数中testExprData = 自己的表达矩阵,测试的表达矩阵换成自己的表达举证就可以了是吗
是的,不好意思 刚刚看见
您好,我输入的表达矩阵为TPM,最终得到的敏感性评分变异比较大,且存在NA值,文献中的敏感性得分基本都在1-10,我甚至达到10^9。请问这是怎么回事?
嗯嗯,这个和其它建模一样,根据特征基因的表达乘以系数得到的评分,你用的TPM数值本身就比较大,而且非正态分布,建议使用log转化的数据
进哥哥,想问您两个问题:1、GDSC2_Expr是所有癌种的细胞系表达信息,如果想做单一癌种,比如肝癌,也是用这个GDSC2-Expr的全癌种信息吗;2、最后得到有统计学意义的药物一共100种,我研究的是肝癌,那呈现结果的时候是只需要把治疗肝癌的药物写出来就可以了对吗?谢谢!!!!
你好,其实最好的做法如您所说,应该以肝癌细胞的数据作为训练集,但是这样往往细胞类型不够,也会导致结果的不稳定,所以一般使用的是所有的;
对于结果呈现,可以进行可视化,比如散点图(类似于基因差异表达的火山图),以及棒棒糖图(变形的条形图),热图(同比基因表达),然后可以将最显著或者最关注的几个列出来
王老师,我只需要索索一个基因但是出现如下报错,怎么处理啊
1 gene identifiers overlap between the supplied expression matrices…
Error in rep(“train”, ncol(trainExprMat)) : invalid ‘times’ argument
In addition: Warning message:
In cbind(trainExprMat, testExprMat) :
number of rows of result is not a multiple of vector length (arg 1)
你是如何处理的 计算药物敏感性需要整个基因表达谱,计算出来之后分期其与目的基因的相关性
如果我需要处理单基因的药物敏感性的话是否可以直接使用单基因的数据就可以了?
不是哟,首先得根据所有基因计算得到敏感性评分,再计算评分与基因表达之间的关系
但是评分算出来有负数,能用么?所以是看越接近0的还是值越小的月敏感
王老师如果我希望进行单个基因的药物敏感性计算呢怎么做呀
你好,现根据这个教程计算得到每个样本的药物敏感性评分,根据所得评分与目的基因做相关性分析
您好,如果想要预测单一药物在泛癌中的敏感性的话,哪儿一部代码选着那个单一的药物呢
您好,这个代码是计算所有样本各个药物的敏感性,如果只需要特定药物,只需要提取出训练集中特定药物数据:GDSC2_Res <- GDSC2_Res[,c("drug")]
您好,我如果在泛癌分析的话,我自己的表达矩阵,是不是基因名为行名,样本名为列名的表达矩阵呀.
github文档示例给出,硼替佐米组预测结果,临床反应为NR的组所得分值更高,合理的解释,最终输出的Drugprediction.CSV中的数值就是细胞意义上的IC50才对。如果按照您所说的是敏感性得分的话,那该结果就与临床反应相反了,明显不对。
您好,我不清楚您说的什么问题?但是就这个R包的文献,作者给出的是:Using the oncoPredict calcphenotype function, imputed sensitivities were generated for each patient against all the drugs in CTRP.
详见:oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data – PMC
欢迎继续讨论,针对这个包我也没做太多了解,只是简单使用
你好,请教一个问题。有些文献里面以IC50为纵坐标,比较高低分险组药物敏感性。这IC50从何而来呢?上述过程得到的结果是数值越大代表越敏感?
是不对的 不是IC50,就是计算出的敏感性得分,应该是文献中错的 IC50越大的话敏感性应该是越低
如果需要 发我你说的文献看看
那请问在绘图的时候可以直接用drugprediction得到的值吗?怎么转换为IC50呢?还是直接在绘图时将纵坐标标为敏感性得分呢?
解决了吗朋友,转化成IC50
没有啊,我就不用IC50了,跟博主说的一样直接描述成敏感值大小
请问一下您是直接将Drugprediction 当做纵坐标了吗?我之前直接将Drugprediction理解为IC50了,结果的出来的结果跟预期是相反的。关于这个问题您有新的发现吗?
对,直接当做纵坐标使用
王博 我看有的文献用的oncoPredict包做的药敏,然后还在图例那里说指数越低,敏感性越高,可以发您看看他们做得对不对吗?
其实我也不清楚,作者和包的说明也没有讲清楚,包说明称结果为药物敏感性,讲道理应该是分数越高,敏感性越高。但原文举的例子好像又确如您说的那个文章中的描述
你好,“因为我们这个是教程,所以我就不读取自己的表达量矩阵了,直截了当的从Genomics of Drug Sensitivity in Cancer (GDSC) 的v2里面随机挑选10个细胞系作为要预测的矩阵。” 如果是自己的数据,能麻烦给写个代码吗?还有说明,谢谢
你好,其实你只要读取数据文件看一下数据结构就知道了,然后直接替换GDSC2_Expr、GDSC2_Res为你们自己的矩阵文件,没有什么其他修改
王博,你好,这里不应该是吧testexp替换成自己的表达矩阵吗
您好,是的, 把 testexp替换成自己的表达矩阵
可以指定某一个肿瘤对应的细胞系及其药敏,来跑这个代码吗?
可以的 你自己提取一下 就是细胞少的情况下数据可能不准确
表达数据格式为TPM或者FPKM,需要进行log处理吗
不是一定得转换 不log可能数据非正态,log其实就是正态化处理 方便后续的一些要求正态的统计方法
我的习惯是都用转化的数据
log以2为底和以10为底区别大吗?
一般2,对于差异分析,需要log2转化,其他的建议都以2
Error in calcPhenotype(trainingExprData = GDSC2_Expr, trainingPtype = GDSC2_Res, :
ERROR: “testExprData” must be a matrix.
王老师,请问这个怎么解决啊,我用的
按照要求,需要将数据框变成matrix,讲你的testExprData数据用as.matrix转换一下
你好!请问表达矩阵应该用TPM还是FPKM呢
都可以的 ,其实就是相关性分析
请问表达矩阵用counts可以吗,unstranded那一列
尽量不要 count没有标准化,存在一定偏差
您好,万分感谢您的教程。我在导入自己的表达谱之后,运行总是提示“Error in tcrossprod(x, y) : 需要数值/复数矩阵/矢量参数”。请问表达谱需要什么条件吗?
您好,没有什么特殊要求 就是数值型表达矩阵,你看一下你的数据里面有没有非字符型。或者不管有没有直接转化:aaa <- apply(aaa,2,as.numeric)
王博您好,根据上面的教程,里面GDSC2_Res的是IC50值,越接近于0越好,但是用自己的表达数据分析得到的文件里面,看你跟另一个人的回复,说只是回归评分,越大代表药物越好,那么根据分析得到的DrugPredictions.csv文件,怎么筛选最好的药物?还有就是想用IC50值画高低风险组药物敏感性对比的箱线图,筛选出来的药物IC50值怎么得到?
您好 不好意思 刚看到留言 809 Cell lines 以及 198 Compounds的数据是在GDSC2_Res.rds文件里,这个算法是根据这些细胞的IC50和表达量作为训练集,得到一个公式能够以基因表达计算药物敏感性,再以你自己的数据作为测试集,根据得到的公式计算每个样本的敏感性。
IC50是细胞上的概念,你最终会得到的数据是肿瘤组织的 这反应的是组织对药物的敏感性
其它还有问题加微信详聊吧,一起讨论
您好,请教以下,请问在做高低风险组的药物敏感性预测时候,自己的表达量矩阵应该怎么制作,变量应该不是细胞系了吧,是不是得把细胞系替换成高风险组病人,蟹蟹
您好 自己的测试集矩阵就是列是样本 行是基因,应该就是你理解的 方法中用到的细胞系表达谱是作为训练集的
您好,我也在做高低风险组的药敏预测。我发现一个有趣的现象:
①使用cellMiner数据库(也是您的网页分享):由于数据库提供“多个细胞系的表达数据”以及“药物对每个细胞系的敏感性数据”,因此我基于这些细胞系的表达数据,使用我构建的模型计算每个细胞系的风险得分。此时我计算(每个细胞系) 风险得分与药敏数据 的相关性:发现p值<0.05的r值均大于0!我就觉得这暗示了随着风险得分的增加,敏感性也增加
②使用本文的GDSC数据:我将我自己数据的病人分成高低风险组,根据不同组的表达数据,计算每个病人对198种药物的敏感性数据。一开始通过热图展示198种药物下不同组病人的敏感性数据,未发现区别。所以我计算了 风险得分与药敏数据 的相关性,发现在所有p<0.05的r值中83个小于0,3个大于0!
为什么不同数据库展示出的药敏相关性南辕北辙呢?是这就是会出现的,还是我的方法哪里有问题呢?
您好 cellminer数据有限,而且仅仅是细胞的,我一般是用的GDSC+oncopredict,这个更加可靠,建议用后者啦
好的收到,感谢进哥,想再确认一下:cellMiner、GDSC+oncpPredict 这两种方法预测出来的都是药物的敏感性数据吗(而不是IC50)?
oncoPredict是敏感性,cellminer下载的其实是药物活性,差不多吧,反正不是IC50,不确定的情况下可以查看相应工具发表的参考文献
王老师,您好,最后输出的DrugPredictions.csv文件有198个药物,表达数据有零点几的,几十的,几百的,请问后面应该怎么筛选,怎么可视化
您好 根据样本进行相关性分析 和你感兴趣的基因或者其它数据 例如risk score等
也可根据目的基因表达或者risk score将所有样本分为高低组 比较两组差异
你好,我已经install了oncoPredict,请问这是R版本的问题吗Error in calcPhenotype(trainingExprData = GDSC2_Expr, trainingPtype = GDSC2_Res, :
could not find function “calcPhenotype”
额 安装好就应该不会是版本问题 你确定加载包了吗? library(oncoPredict)
感谢,我已经解决了这个问题并且得到了很棒的结果,你的教程真的很棒?
请问同年怎么解决的呢?我也遇到同样的问题,是R版本的问题吗?
有同学说也遇到了这个问题,可以试试用bioconductor装一下这个包
王老师,您好,请问最后输出的DrugPredictions.csv文件,这个矩阵数值指的是每个样本对应的不同药物的IC50值吗
当然不是,这个数值是通过回归计算计算出的一个评分,根据评分大小反应敏感性高低
再麻烦问您一下,那这里面的数值越大代表药物敏感性越高,这个样本越适用于这种药物吗
是的,按照工具说明是这样
您好,请问DrugPredictions文件里出现好几个药物是负值是怎么回事?
进哥,你在视频里不是说score的值越大,敏感性越低吗,到底应该怎么看score的大小呀
请问药物名称后边的数字代表什么意思,例如Cisplatin_1005
这个是GDSC数据库中的Drug_ID:GDSC-download