CellMiner数据库,主要是通过国家癌症研究所癌症研究中心(NCI)所列出的60种癌细胞为基础而建立的。该数据库最初发表于2009年,后于2012年在Cancer Research杂志上进行了更新,题目为“CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set”。大家后期在使用该数据库记得应用相关文献。

NCI-60细胞系是目前使用最广泛的用于抗癌药物测试的癌细胞样本群。大家可以通过它查询到 NCI-60细胞系中已确认的22379个基因,以及20503个已分析的化合物的数据(包括多种已获美国食品和药物监督局批准的药物,以及临床试验中的药物分子)。
下面,我们来看下相关数据的下载和药物敏感性分析过程。
1.CellMiner数据库的使用
1. 进入CellMiner数据库主页(https://discover.nci.nih.gov/cellminer/home.do);
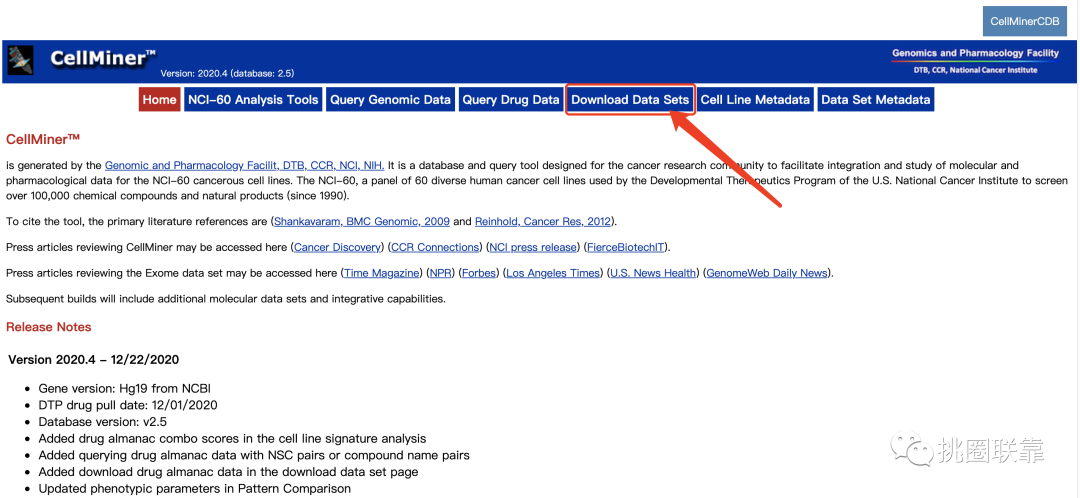
2. 点击Download Data Sets,进入数据下载界面;在下载界面中,可以看到两个不同的板块,分别是原始数据Raw Data Set和经过处理后的数据Processed Data Set;

3. 在此,我们直接选择经过处理后的数据Processed Data Set,勾选RNA表达数据(RNA: RNA-seq)和药物数据(Compound activity: DTP NCI-60);
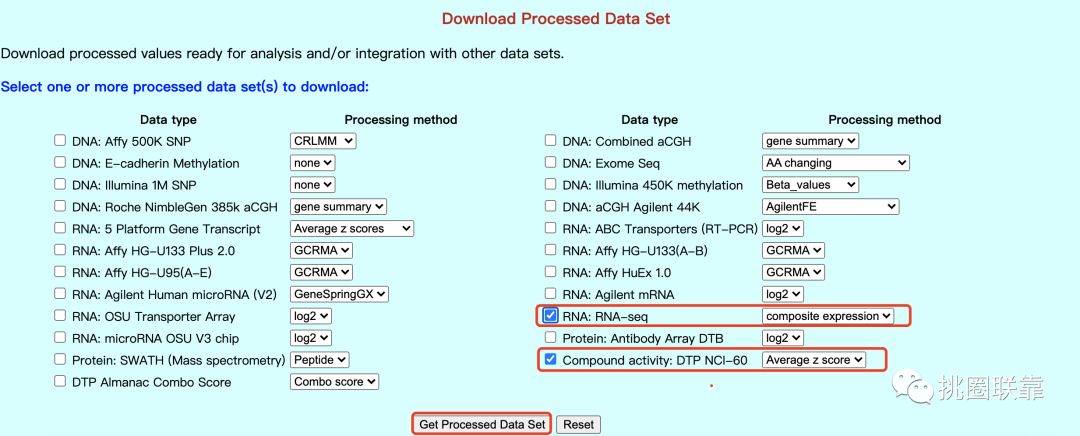
4. 点击按钮Get Processed Set,进入下载界面,点击即可保存;

5.下载完成后,将其放到工作目录下,解压,并分别提取其中的Excel文件放于当前工作目录下;

接下来,我们需要对下载得到两个数据文件进行整理,以用于后续的药物敏感性评估。
2.药物数据的准备
2.1 读取药物相关数据
library(readxl)
rt1 <- read_excel(path = "DTP_NCI60_ZSCORE.xls", skip = 7)
首先,使用readxl包中的read_excel()函数,读取药物相关的数据。由于前7行为注释信息,因此使用参数skip进行跳过前7行。
colnames(rt1) <- rt1[1,]
rt1 <- rt1[-1,-c(67,68)]
同时,将第一行作为列名,并去除末尾两列其余信息。
2.2 筛选药物标准
table(rt1$`FDA status`)使用table()函数药物标准,结果显示,其中75种经过了临床试验,188种经过FDA批准。
rt1 <- rt1[rt1$`FDA status` %in% c("FDA approved", "Clinical trial"),]
rt1 <- rt1[,-c(1, 3:6)]
write.table(rt1, file = "drug.txt",sep = "\t",row.names = F,quote = F)
为了保证分析结果的可靠性,选取经过临床试验(Clinical trial)和FDA批准(FDA approved)的药物结果。当然,你也可以选择将所有的药物进行保留。最终,一共得到263个药物结果,并将其保存为txt文件用于后续分析。
3. 基因表达数据的准备
rt2 <- read_excel(path = "RNA__RNA_seq_composite_expression.xls", skip = 9)
colnames(rt2) <- rt2[1,]rt2 <- rt2[-1,-c(2:6)]
write.table(rt2, file = "geneExp.txt",sep = "\t",row.names = F,quote = F)
同样的,读取表达数据,并对其进行整理和保存,用于后续的分析。
4. 药物敏感性分析
rm(list = ls())4.1 引用包
library(impute)
library(limma)
首先,加载分析使用的R包,包括impute包和limma包。
4.2 读取药物输入文件
rt <- read.table("drug.txt",sep="\t",header=T,check.names=F)
rt <- as.matrix(rt)rownames(rt) <- rt[,1]
drug <- rt[,2:ncol(rt)]
dimnames <- list(rownames(drug),colnames(drug))
data <- matrix(as.numeric(as.matrix(drug)),nrow=nrow(drug),dimnames=dimnames)
首先,读取前面保存的药物敏感性结果,设置相应的行名,并将其转换为矩阵形式。
mat <- impute.knn(data)
drug <- mat$data
drug <- avereps(drug)
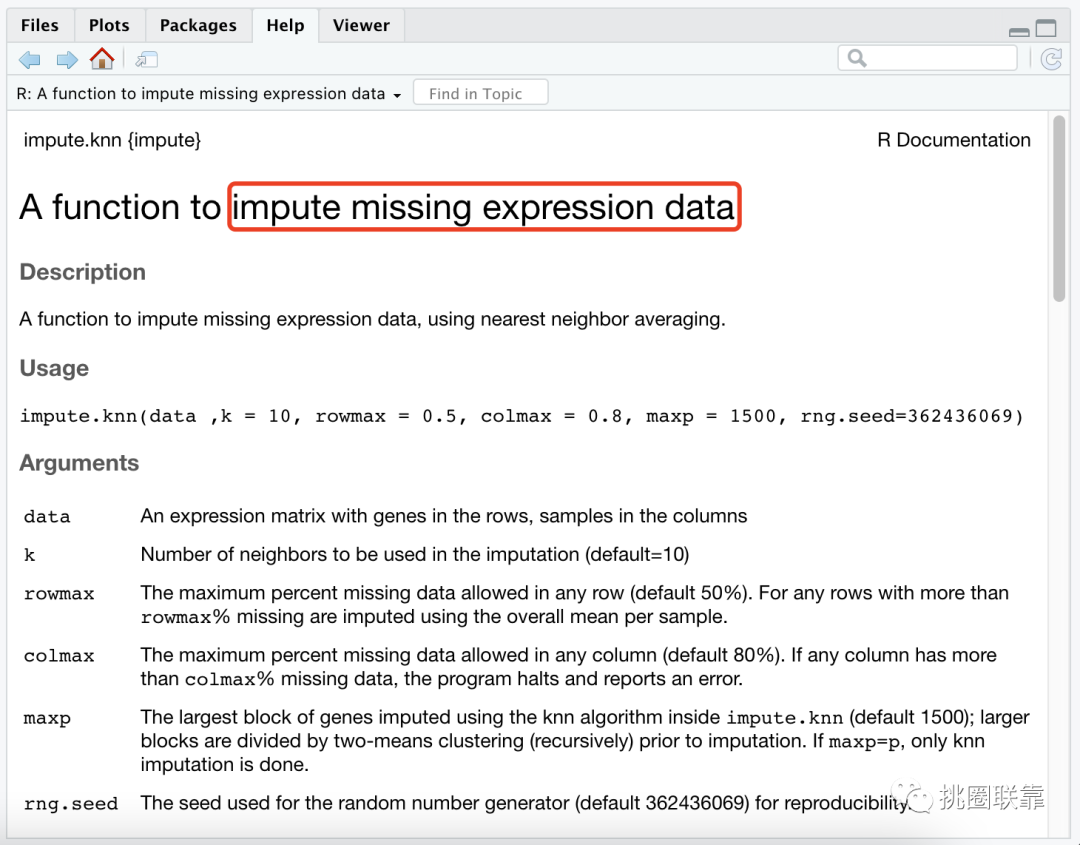
考虑到药物敏感性数据中存在部分NA缺失值,通过impute.knn()函数来评估并补齐药物数据。其中,impute.knn()函数是一个使用最近邻平均来估算缺少的表达式数据的函数。
4.3 读取表达输入文件
exp <- read.table("geneExp.txt", sep="\t", header=T, row.names = 1, check.names=F)
dim(exp)exp[1:4, 1:4]
同时,读取整理完成的NCI-60细胞系中基因表达情况。结果显示:其中包含了60种不同肿瘤细胞系,23805个基因的表达情况。
4.4 提取特定基因表达
gene <- read.table("gene.txt",sep="\t",header=F,check.names=F)
genelist <- as.vector(gene[,1])
genelist
将提前准备的目标基因列表进行读取;结果显示,包括FANCD2,BRCA1,ABCC1,TP53,EGFR基因。
genelist <- gsub(" ","",genelist)
genelist <- intersect(genelist,row.names(exp))
exp <- exp[genelist,]
4.5 药物敏感性计算
首先,新建一个空的数据框,用于保存后续分析结果。
outTab <- data.frame()
for(Gene in row.names(exp)){
x <- as.numeric(exp[Gene,])
#对药物循环
for(Drug in row.names(drug)){
y <- as.numeric(drug[Drug,])
corT <- cor.test(x,y,method="pearson")
cor <- corT$estimate
pvalue <- corT$p.value
if(pvalue < 0.01){
outVector <- cbind(Gene,Drug,cor,pvalue)
outTab <- rbind(outTab,outVector)
}
}
}随后,使用for循环,分别计算每个基因表达与不同药物之间的Pearson相关系数。根据P值<0.01为界,对分析结果进行筛选,并将结果保存给变量outTab。结果显示:最终得到了63个相关性分析结果。
outTab <- outTab[order(as.numeric(as.vector(outTab$pvalue))),]
write.table(outTab, file="drugCor.txt", sep="\t", row.names=F, quote=F)
最后,将相关性分析结果进行输出保存,后续进行可视化
您好,后面可视化的代码可以分享一下吗?在其他地方找了很多都跑不出来,非常感谢!
指的是相关性分析的结果吗?我B站应该有类似的代码分享
好的,请问一下您在B站的账号是什么呢?我去学习一下
你好,我想请教一下:install.packages(“impute”)
Error: package ‘impute’ is not available
In addition: Warning messages:
1: could not retrieve available packages for url “https://cran.rstudio.com/src/contrib”
2: could not retrieve available packages for url “https://cran.rstudio.com/bin/windows/contrib/4.2”
3: curl: (23) Failure writing output to destination
4: package ‘impute’ is not available
5: failed to retrieve ‘https://rstudio-buildtools.s3.amazonaws.com/renv/mran/packages.rds’ [error code 23]
出现这样的结果,安装不了”impute”包怎么解
不知道你有没有安装好,这个包是在bioconductor上的https://bioconductor.org/packages/release/bioc/html/impute.html
如是安装:BiocManager::install(“impute”)
大哥,能不能分享一下你跑了出来的整个R包?非常感谢您!
结果是这样的:
BiocManager::install(“impute”)
Error: unexpected input in “BiocManager::install(“”
解决了吧?引号问题,用的中文的
进哥,目前“impute”包已解决,目前又来麻烦你了,我在对结果进行可视化过程中,即箱式图绘制过程中,出现了如下错误:plotList_2 corPlotNum if (nrow(outTab) < corPlotNum) {
+ corPlotNum for (i in 1:corPlotNum) {
+ Gene <- outTab[i, 1]
+ Drug <- outTab[i, 2]
+ x <- as.numeric(exp[Gene, ])
+ y <- as.numeric(drug[Drug, ])
+ df1 <- as.data.frame(cbind(x, y))
+ colnames(df1)[2] <- "IC50"
+ df1$group median(df1$x), “high”, “low”)
+ compaired <- list(c("low", "high"))
+ p1 <- ggboxplot(df1,
+ x = "group",
+ y = "IC50",
+ fill = "group",
+ palette = c("#00CCBF", "#FF5F5D"),
+ add = "jitter",
+ size = 0.5,
+ xlab = paste0("The expression_of_ ", Gene),
+ ylab = paste0("IC50_of_", Drug)) +
+ stat_compare_means(comparisons = compaired,
+ method = "wilcox.test",
+ symnum.args = list(cutpoints = c(0, 0.001, 0.01, 0.05, 1),
+ symbols = c("***", "**", "*", "ns")))
+ plotList_2[[i]] <- p1
+ }
Error in names(x) <- value :
'names' attribute [2] must be the same length as the vector [1],想请教一下您!
你好,还是微信讨论吧,加微信好了
您好,请问这个网站预测的是哪一类肿瘤细胞的敏感性呢?如何理解其预测原理?
您好 这个我也不清楚,您看一下原文那个参考文献学习一下吧
进哥你好,打扰请教一下:
我在运行这个代码的第四部分的时候, 采用for循环进行药物敏感性计算,一直报错:Error in if (pvalue < 0.01) { : missing value where TRUE/FALSE needed,后反复检查,发现对drug矩阵进行row.names(drug)操作的时候,就会陷入巨大的计算量,一直进入卡死状态,后来我发现只要对这个drug矩阵的行名进行任何操作都会进入卡死状态,如果把行名变成第一列之后,对第一列进行读取还是一样的结果,各种检查都不知道为什么,请问进哥是否遇到过这类问题?
麻烦啦。
你好 如果还没解决,方便的话加我微信,我看一下数据
你好,我按照上诉代码跑结果,最后一步时出现了问题,请问王老师能否抽空回复一下
最后一步画barplot组合图时,p值全是ns,反馈结果如下:
> ggarrange(plotlist=plotList_2,nrow=nrow,ncol=ncol)
Warning messages:
1: In wilcox.test.default(c(-1.61, -1.49, -1.54, 0.19, 0.9, 0.95, -0.29, :
无法精確計算带连结的p值
2: In wilcox.test.default(c(-2.81, -1.14, -0.67, -0.57, -0.5, 0, -2, :
无法精確計算带连结的p值
3: In wilcox.test.default(c(-1.61, -1.61, -1.13, -0.29, 0.06, -1.37, :
无法精確計算带连结的p值
4: In wilcox.test.default(c(-0.31, -1.09, -0.44, -0.06, 0.06, 0.11, :
无法精確計算带连结的p值
5: In wilcox.test.default(c(-0.44, -0.55, -1.44, 0.5, 0.1, 0.42, -1.93, :
无法精確計算带连结的p值
6: In wilcox.test.default(c(-1.66, -1.77, 0.96, -0.39, -0.42, -0.54, :
无法精確計算带连结的p值
7: In wilcox.test.default(c(-0.8, -0.8, -0.7, -0.8, -0.18, -0.75, -0.8, :
无法精確計算带连结的p值
8: In wilcox.test.default(c(-1.15, -1.29, -0.73, -0.64, 0.29, -0.59, :
无法精確計算带连结的p值
9: In wilcox.test.default(c(-1.2, -1.39, -0.27, -0.69, 0.22, 0.66, :
无法精確計算带连结的p值
不好意思老师,才发现后面的代码是别处复制过来的
plotList_2 <- list()
corPlotNum <- 16
if(nrow(outTab)<corPlotNum){
corPlotNum=nrow(outTab)
}
for(i in 1:corPlotNum){
Gene <- outTab[i,1]
Drug <- outTab[i,2]
x <- as.numeric(exp[Gene,])
y <- as.numeric(drug[Drug,])
df1 <- as.data.frame(cbind(x,y))
colnames(df1)[2] <- "IC50"
df1$group median(df1$x), “high”, “low”)
compaired <- list(c("low", "high"))
p1 <- ggboxplot(df1,
x = "group", y = "IC50",
fill = "group", palette = c("#00AFBB", "#E7B800"),
add = "jitter", size = 0.5,
xlab = paste0("The expression of ", Gene),
ylab = paste0("IC50 of ", Drug)) +
stat_compare_means(comparisons = compaired,
method = "wilcox.test", #设置统计方法
symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "ns")))
plotList_2[[i]]=p1
}
nrow <- ceiling(sqrt(corPlotNum))
ncol <- ceiling(corPlotNum/nrow)
ggarrange(plotlist=plotList_1,nrow=nrow,ncol=ncol)
ggarrange(plotlist=plotList_2,nrow=nrow,ncol=ncol)
直接加我微信吧 微信交流 需要你绘图的文件 这样我方便修改
您好,我用的TCGA数据库通过分析将患者分成高低风险两组,对比两组的药敏分析,我要如何准备数据呢
您好,就是分别计算两组的风险评分与药物敏感性的相关性,药物相关数据参考文中的 你的数据就是高低风险组各个样本的风险评分
想问问具体代码可以指点下嘛,用的TCGA数据库通过分析将患者分成高低风险两组,对比两组的药敏分析
我先解释一下计算过程 不懂加微信讨论
你应该已经由你的训练数据得到一个计算Risk score的signatures和计算公式 将此公式应用于cellminer中基因表达数据集,由此得到各种细胞的risk score 再计算这个score与药物敏感性数据的相关性
您好,请问方便加您微信吗?我也是分组后,关于比较两组之间药物敏感性的问题想请教下您
不好意思 前两天忙备课高校教资考试,可以的 导航栏我的简历有手机号和微信
运行:
for(Gene in row.names(exp)){
x <- as.numeric(exp[Gene,])
#对药物循环
for(Drug in row.names(drug)){
y <- as.numeric(drug[Drug,])
corT <- cor.test(x,y,method="pearson")
cor <- corT$estimate
pvalue <- corT$p.value
if(pvalue < 0.05){
outVector <- cbind(Gene,Drug,cor,pvalue)
outTab <- rbind(outTab,outVector)
}
}
}
时出现:
Error in if (pvalue < 0.05) { : missing value where TRUE/FALSE needed
In addition: Warning message:
In cor(x, y) : 标准差为零,
这个是怎么回事呢
你好,如报错信息,标准差为0,不能做相关性分析,因为有些列数据都一样,导致sd=0,
可以加一个if判断
不清楚加我微信交流
太感谢了,跑通了
同问怎么加if判断呢,我也是报了同样的错
请参考如下修改:
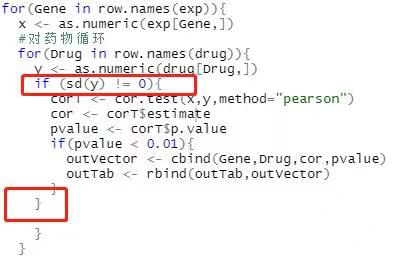
具体怎么加if判断呢,我也是报了同样的错
请参考如下修改:
具体怎么加if判断呢,我也是报了同样的错
不好意思 之前链接没有加上,请参考如下修改:

你好,Error in impute.knn(data) : a column has more than 80 % missing values!
这个错误怎么解决呢?补齐数据补不上,呜呜
你好,正如报错提示,因为缺失值太多,knn法不能准确地补充缺失值,这个时候你需要先把缺失值过多的基因删除,比如我会把缺失值多于60%的基因删除
可以用Na_ratio <- apply(data,2,function(x) sum(is.na(x))/length(x))>0.6计算得到各列确实值比例,然后data[-Na_ratio]即可
大佬好,以下是我练习的流程,小白一个
#开始药物敏感性分析
rm(list = ls())
library(impute)
library(limma)
#读取药物输入文件 读取前面保存的药物敏感性结果,设置相应的行名,并将其转换为矩阵形式。
rt <- read.table("drug.txt",sep="\t",header=T,check.names=F,quote="")
#因为报错和有些数据缺失,我加了个quote="" 不知问题大不大?
rt <- as.matrix(rt)
rownames(rt) <- rt[,1]
drug <- rt[,2:ncol(rt)]
dimnames <- list(rownames(drug),colnames(drug))
data <- matrix(as.numeric(as.matrix(drug)),nrow=nrow(drug),dimnames=dimnames)
#这里也因为转换的原因报错了NAs introduced by coercion
#这样加可以吗?
Na_ratio 0.6
data[-Na_ratio]
#通过impute.knn()函数来评估并补齐药物数据
mat 0.6的一列后,还是出现同样的报错
初学者,问题可能很蠢,求大佬解答
data <- matrix(as.numeric(as.matrix(drug)),nrow=nrow(drug),dimnames=dimnames)
Na_ratio 0.6
data[-Na_ratio]
更正一下
Na_ratio 0.6 出不来
不好意思 前两天太忙 没时间回复网站留言,可以的话你直接加微信交流吧
进哥您好,我也遇到了和上面这位一样的问题,data[-Na_ratio]后依然是报错提示缺失值太多,想问一下该怎么解决
还没解决的话 加我微信 我看看 估计是你操作有误
进哥您好,我也遇到了和上面这位一样的问题,data[-Na_ratio]后依然是报错提示缺失值太多,想问一下该怎么解决
您好 可以加微信看看你的数据 现在我也给不出解答
你好,请问你这个问题解决了吗?我也遇到了和你同样的问题。
你好,我按照上诉代码不能完全跑出上述结果,出现的问题:感觉R代码不全,不知能否帮忙指导:
在第4.5 药物敏感性计算时,运行
outTab <- data.frame()
for(Gene in row.names(exp)){
x <- as.numeric(exp[Gene,])
#对药物循环
for(Drug in row.names(drug)){
y <- as.numeric(drug[Drug,])
corT <- cor.test(x,y,method="pearson")
cor <- corT$estimate
pvalue <- corT$p.value
if(pvalue < 0.01){
outVector <- cbind(Gene,Drug,cor,pvalue)
outTab <- rbind(outTab,outVector)
}
}
出现该错误:
Error in rbind(outTab, outVector) :
cannot coerce type 'closure' to vector of type 'list'
您好,看一下中断时的位置,也就是drug是哪个?单独运行查看原因,推测是由于某个药物数据较多缺失值,搞不定加我微信远程看看
您好,想请教一个问题:cellminer中做出来的药物敏感性拟合图,为什么纵坐标IC50,但基因表达与ic50正相关时代表基因对此药物敏感度高? IC50 不是越低药物才越有效,细胞才越敏感么?
您好,您为什么说纵坐标是IC50,它的单位在数据库中是个Z score,反映的就是药物敏感性指数
感谢您的回复。我是刚开始看生信文章,有篇文章作者的Supplementary Figure 3 纵坐标是ic50,给了我误解…doi: 10.3389/fonc.2022.830174, 再次感谢您的回复