RNAhybrid的介绍
RNAhybrid是Behmsmeier M等基于miRNA和靶基因二聚体二级结构开发的miRNA靶基因预测软件。RNAhybrid预测算法禁止分子内、miRNA分子间及靶基因间形成二聚体,根据miRNA和靶基因间结合能探测最佳的靶位点。尽管随着靶基因序列长度增加,运算复杂度也相应增加,但RNAhybrid和其它RNA二级结构预测软件诸如mfold, RNAfold, RNAcofold和pairfold相比,仍具有明显的速度优势。此外,RNAhybrid允许用户自定义自由能阈值及p值,也允许用户设置杂交位点的偏向,如杂交位点必须包含miRNA 5’端2-7nt等。
1.RNAhybrid的下载与安装
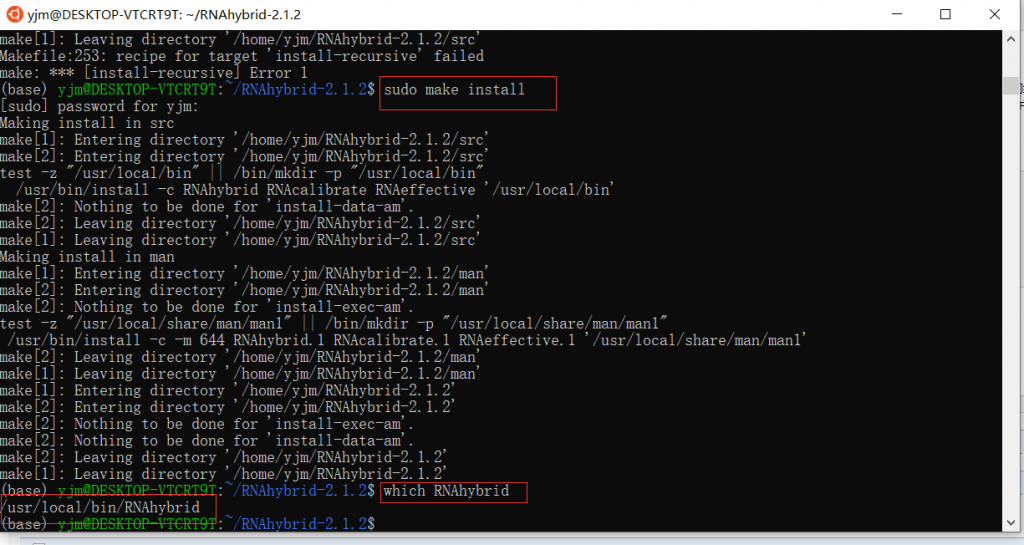
wget https://bibiserv.cebitec.uni-bielefeld.de/applications/rnahybrid/resources/downloads/RNAhybrid-2.1.2.tar.gz tar -xzvf RNAhybrid-2.1.2.tar.gz cd /path/to/ RNAhybrid-2.1.2 ./configure sudo make #这里尽量使用管理员模式,不然容易出错 sudo make install
验证是否安装成功,可以输入which RNAhybrid,如显示地址,则安装成功,以下是用win10下的WSL下的ubuntu做的示范:
2.输入文件的准备
1.target sequence(s)
This contains one or more sequences that are used by RNAhybrid to hybridize the miRNA(s) on. RNAhybrid uses all this sequences to find minimal free energy hybridisations between miRNA(s) and target sequence(s). Sequences should be in RNA.fasta format but RNAhybrid can also use DNA.fasta files. A single Sequences one can use can contain up to 50000 basepairs.
这里的target sequence用的是从circbase下载的人的circRNA的fasta文件,具体下载方法参考我这篇博客https://www.cnblogs.com/yanjiamin/p/12057362.html
2.miRNA sequence(s)
contains one or more micro RNA(s) that RNAhybrid uses to hybridize with the RNA sequences and to find the minimal free energy hybridization. A single micro RNA sequence can contain up to 2000 basepairs.
这里的miRNA sequence用的是从miRbase下载的成熟的人的miRNA的fasta文件,具体下载方法参考我这篇博客https://www.cnblogs.com/yanjiamin/p/12057362.html
3.RNAhybrid的使用
Usage: RNAhybrid [options] [target sequence] [query sequence].
options:
-b <number of hits per target> #意思是一个miRNA和一个target sequence的某一段序列匹配情况最多列出几次,比如一个miRNA和一个target sequence的某一段序列匹配存在多种情况,则-b 1就是列出最优的匹配情况,一般选1就比较好。这个最终得到的数目也与<energy cut-off>的设定值有关。
-c compact output #使用这个参数,每一个匹配只会显示一行输出。如果只想知道结果是否与RNAhybrid校准的结果相同,建议使用这个参数。
-d <xi>,<theta> #位置和形状参数
-f helix constraint #
-h help
-m <max targetlength>
-n <max query length>
-u <max internal loop size (per side)> #内部成环的错配碱基的个数,使用-u 0,将得到完全没有错配碱基内部成环的结构。
-v <max bulge loop size> #internal loop是两条链都没有结合位点的内部环,而bulge loop是某一条上多出的碱基的突出
-e <energy cut-off> #两条序列匹配的最低自由能,先设置 -e -30看看效果。
-p <p-value cut-off>
-s (3utr_fly|3utr_worm|3utr_human) #用于极值分布参数的快速估计,你可以选择nothing,3utr_fly, 3utr_worm和3utr_human来更好的匹配这些物种。你不能同时使用helix constrain和approximate p-value这两个参数。
-g (ps|png|jpg|all) #图片输出的格式,有ps,png,jpg或者all四个选项
-t <target file> #fasta格式的target gene文件
-q <query file> #fasta格式的miRNA文件
Either a target file has to be given (FASTA format)
or one target sequence directly.
Either a query file has to be given (FASTA format)
or one query sequence directly.
The helix constraint format is “from,to”, eg. -f 2,7 forces
structures to have a helix from position 2 to 7 with respect to the query.
<xi> and <theta> are the position and shape parameters, respectively,
of the extreme value distribution assumed for p-value calculation.
If omitted, they are estimated from the maximal duplex energy of the query.
In that case, a data set name has to be given with the -s flag.
PS graphical output not supported.
PNG and JPG graphical output not supported.
| Name | Description |
|---|---|
| helix constraint from | Forces all structures to have a helix from position a to position b in respect to the query. The first base has position 1. The parameter “Helix constrain from” has to be lower or equal to the parameter “Helix constraint to”. You can not use Helix constraint and approximate p-values at the same time. |
| hits per target | This Parameter defines how many hits are shown by RNAhybrid. The hits are shown by increasing minimal free energy ( the lower the energy the better the result) |
| Compact output | When this parameter is used RNAhybrid gives you only one line of outputinstead of the whole output it normally generates. |
| Generate graphics | Generates a graphical representation of the output in jpg, png and ps format, if less than 6 hits choosen. If RNAhybrid breaks with an unexpected error, it is often a good idea not to enable the graphical representation generation. |
| Max internal loop length | The maximal number of unpaired nucleotides in either side of an internal loop. |
| energy Threshold | Shows the hits with all minimal free energy’s lower then the threshold (the lower the result the better). The value has to be lower or equal to zero.Notice that the output only shows the results that exceed the energy threshold or the maximal hits per target. |
| Max bulge loop length | the maximal number of unpaired nucleotides in a bulge loop. |
| No G:U in seed | If you click on this you choose weather their are no G:U bindings allowed in the seed or not. This parameter can only be chosen if you also use the parameters “Helix constraint from” and “Helix constraint to”. |
| helix constraint to | see helix constraint this is position b you have to use both parameters to use Helix constraints. |
| approximate p-value | Used for a quick estimate of extreme value distribution Parameters. You can choose between nothing, 3utr_fly, 3utr_worm and 3utr_human for better equitation within these species. You can not use Helix constraint and approximate p-values at the same time. |
4.RNAhybrid进行人miRNA的靶位点预测的条件
1.miRNA的第8到12个碱基和circRNA的必须是完全配对的,这里需要设置的参数是-f helix constraint,也就是设置-f 8,12
2.是指上下两条链都错配形成的错配环,这种错配环中任何一条链的错配碱基不能超过1个,这里需要设置的参数是-u <max internal loop size (per side)> ,也就是设置-u 1
3.突出环即一条链多出了一个碱基的突出,这种突出环最多突出一个碱基,这里需要设置的参数是-v <max bulge loop size> ,也就是设置-v 1
4.允许G:U配对,默认的参数是允许G:U配对,你也可以设置no G:U in seeds来设置不允许G:U配对
5.末端未配对的突出不能超过两个碱基
6.不允许存在连续3个碱基的错配
7.总数不超过4个碱基的错配
RNAhybrid -g jpg -b 1 -e -20 -f 8,12 -u 1 -v 1 -s 3utr_human -t SFTSV_24vscontrol_DEcircBase.fa -q hsa_miRNA.fa>SFTSV_24bscontrol_circRNA_miRNA_RNAhybrid #输出会直接打印在终端里,所以建议你在终端以 “>" 输出保存为一个文件
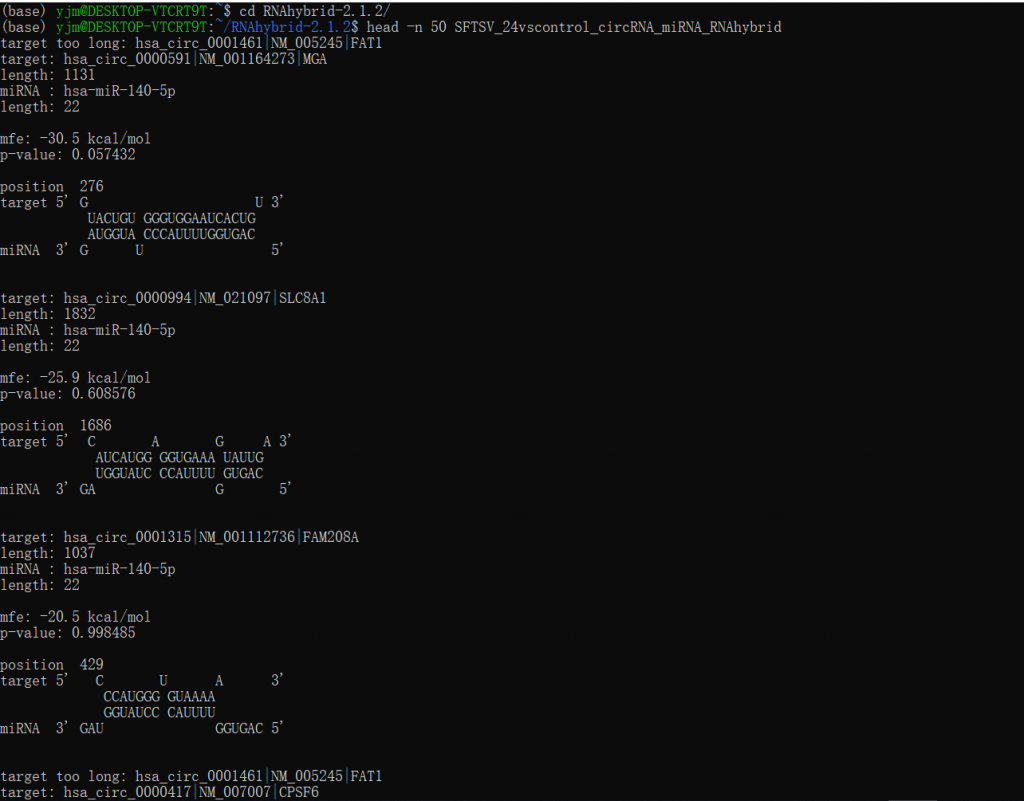
当然,网站提供了win版本离线软件,按照说明加入环境变量,然后在CMD中同上运行
大佬你好!请问你有做过有这些预测mirna靶基因的数据库(miranda,targetscan等等)来预测tsRNA的靶基因吗?感谢抽空回答!
看到你加我微信了 最近有点忙 网站回复不及时 就如我微信所说,可以利用基于序列预测靶基因的工具 比如RNA22、hybrid等
RNA22预测miRNA靶基因
miRNA结合位点预测软件RNAhybrid的使用教程