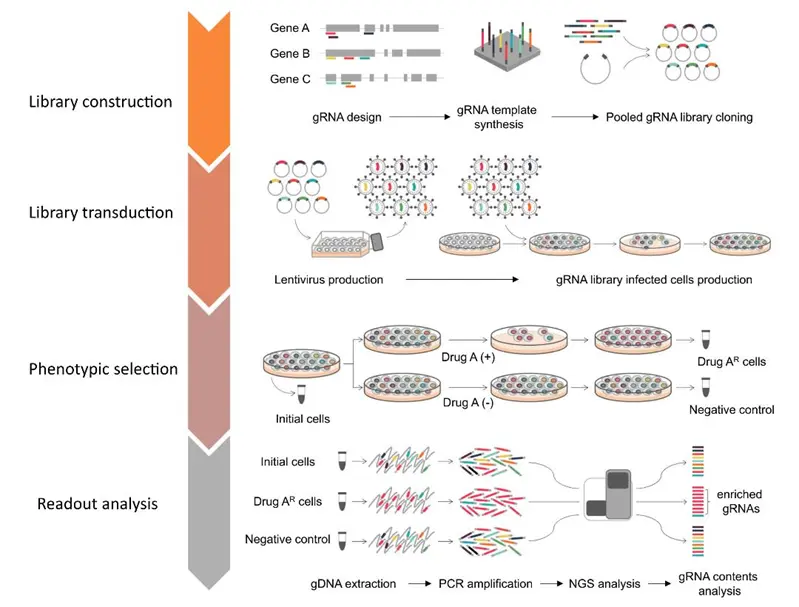
CRISPR/Cas9筛选流程:
1. 文库构建:针对某个物种,每个基因设计3个或以上的sgRNA,高通量合成sgRNA,然后把合成的sgRNA克隆到慢病毒载体中。
2. 慢病毒转导:包装GeCKO慢病毒文库,并以低MOI(一般标准<0.3)感染靶细胞,保证一个病毒颗粒感染一个细胞,筛选同时表达sgRNA和Cas9的细胞;每个细胞都只会敲除自身携带的sgRNA对应的一个基因,细胞文库中包含的细胞的数量一般为细胞文库中全部sgRNA数量的100-1000倍。
3. 表型筛选:将全基因组敲除细胞库分成两份,其中一份作为实验组施加筛选压力,如:病毒侵染,药物治疗等;另一份作为对照组。根据耐药性、增殖能力、存活能力等表型筛选细胞。
4. 候选基因的分析:将实验组和对照组的细胞分别抽提基因组,PCR扩增sgRNA片段,高通量测序,并进行生物信息学分析。
根据筛选目的的不同,分为阳性筛选和阴性筛选。阳性筛选是对已成功整合sgRNA的细胞文库施加一定的筛选压力,仅使少数目的表型的细胞能够存活,达到富集关键基因的目的。阴性筛选与之相反,存活的细胞并不是目的表型细胞,需要比较不同时间点sgRNA的丰度找出差异sgRNA来确定关键基因,阴性筛选可以鉴定出引起细胞某些功能缺失的基因,如筛选时间较长,可以筛选到细胞生存所必需的基因。
个人理解:阳性、阴性筛选或者正向、负向筛选听起来容易混淆,其实在英文中就2个词,positive selection和negative selection,无论怎么选择,最后都是看sgRNA的计数情况,然后根据实验目的确定候选基因。如果最后选出的细胞内富集的sgRNA所靶向的基因就是我们要的目的基因,那么这种选择我们称其为positive selection。相反,如果最后富集的sgRNA并不是我们实验想要的,我们通过比较不同时间点的sgRNA变化进而推断候选基因,这样的选择称之为negative selection。举个例子,我们要筛选细胞生长的必需基因,如果把这个基因敲除了(这个基因由其对应的sgRNA作为标记,每个细胞内都敲除了一个基因),那么细胞肯定不能存活了,对应的sgRNA肯定也不会富集。所以存活的细胞意味着没有敲除必需基因,随着细胞的增值,整合到其基因组上的sgRNA大量富集,我们最终测到了这些sgRNA,但其靶向的基因敲除了并不影响细胞生长,所以这些基因不是目的基因。因此我们比较不同时间点sgRNA的消耗情况,那些sgRNA明显减少的,其靶向的基因才是必需基因。
那么什么情况下是positive selection?我们知道,这两者是反的,所以假如我们想筛选细胞生长的抑制基因(和必需基因相反),这种情况就属于positive selection(和negative selection相反)。如果敲除了抑制基因,显然细胞可以正常增值,抑制基因对应的sgRNA也能不断富集,这两个方向是相同的。
如果富集sgRNA靶向的基因就是目的候选基因,则为positive selection,否则为negative selection。
阳性筛选实例:
①筛选炭疽毒素使细胞中毒所必需的宿主基因
由于毒素的强选择性压力,大多数细胞都会死亡,只有少量的细胞存活和增殖,这些存活的细胞内富集了大量sgRNA,而这些sgRNA靶向的基因是被敲除的,这些基因就是使细胞中毒所必需的宿主基因,正是由于这些目的基因被敲除,细胞才得以存活。
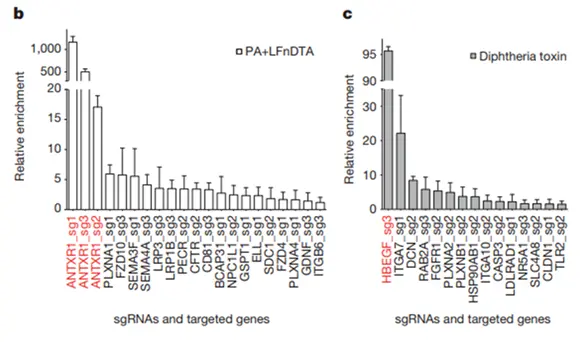
Zhou等(2014)利用敲除文库筛选,在初步确定的291个基因中成功鉴定出炭疽和白喉毒素致细胞中毒所必需的宿主基因,并通过功能验证得到了证实。
②筛选药物敏感基因
细胞若含有药物敏感基因将失去抗药性。若敲除了敏感基因,则可以存活,最终富集的sgRNA正是实验目的需要的sgRNA。
阴性筛选实例:
①鉴定细胞生长必需的基因
一个时间段内的连续生长,减少的细胞中往往携带靶向细胞增殖所必需基因的sgRNA。这些基因可以通过比较每个sgRNA的相对频率来找到。阴性筛选的一个重要应用是鉴定癌细胞生长所必需的基因,而这些基因可能成为治疗癌症的新靶标。
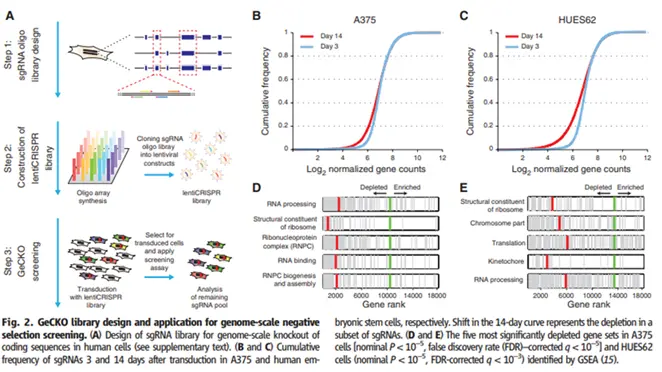
2014年,Shalem等首次利用GeCKO文库鉴定出人黑色素瘤细胞和多能干细胞存活的关键基因,研究结果与RNAi筛选结果高度一致。
②筛选药物抗性基因
若敲除了抗药性基因,细胞在药物压力下不能存活,存活的细胞内并没有敲除目的基因,最终富集的sgRNA不是目的sgRNA,需要通过与对照组比较sgRNA消耗情况(或不同时间点的sgRNA丰度)来确定目的基因。
生物信息学分析:
1. 测序数据质控,去除低质量的reads,使用clean reads data进行后续分析。
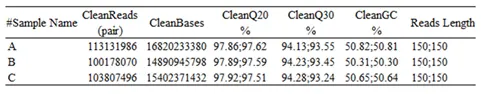
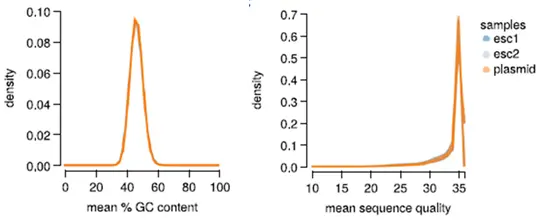
2.比对分析:将测序数据中拼接reads与sgRNA 文库序列比对,并对sgRNA 文库中完全匹配的sgRNA 数目、丢失sgRNA、基尼指数等进行统计。
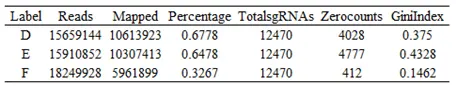
•Reads: Total number reads in the fastq file. (Recommended: 100~300 times the number of sgRNAs)
• Mapped: Reads that can be mapped to gRNA library
• Percentage: The percentage of mapped reads (Recommended: at least 60%)
• TotalsgRNAs: The number of sgRNAs in the library
• ZeroCounts: The number of sgRNA with 0 read counts(Recommended: no more than 1%)
• GiniIndex: The Gini Index of the read count distribution. Gini index can be used to measure the evenness of the read counts, and a smaller value means a more even distribution of the read counts. (Recommended: around 0.1 for plasmid or initial state samples, and around 0.2-0.3 for negative selection samples )
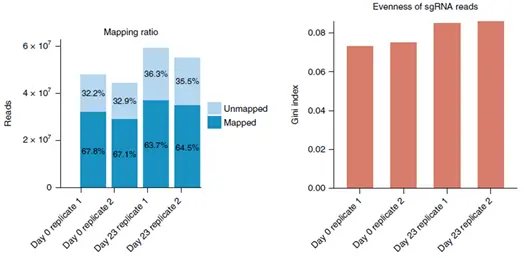
3.sgRNA 及基因的read counts 统计
CRISPR全基因组筛选的主要内容便是统计sgRNA在不同样本间的消耗和富集情况,进而推断候选基因。
4.主成分分析和样品相关性聚类分析
有助于考察样本间的相似性和差异,评估实验设计的合理性以及对后续数据分析起到一定指导作用。对于有重复的实验来说,样本同一处理不同重复应该尽可能聚在一起,若存在个别重复样本明显偏离,可以考虑剔除该样本。样品聚类还可用于判断数据处理中是否剔除了批次效应。
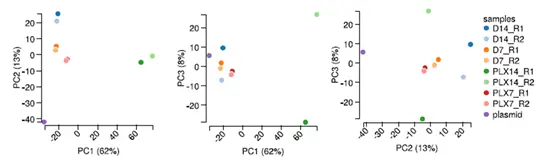
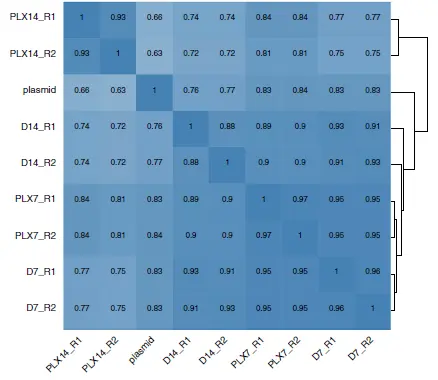
5.候选基因筛选
MAGeCK-RRA算法根据测序结果中sgRNA的富集情况产生对应基因位点的RRA得分,RRA得分代表基因的必要性,RRA得分越小表明其重要性越高。MAGeCK返回RRA lo value(neg|score、pos|score)检验统计显著性,并据此对基因进行了排序,按照实验目的的不同,候选基因筛选可分为正向筛选和负向筛选,筛选指标一般参考lfc(log2 fold change)和FDR(adjusted-pvalue),比如:根据lfc<-2且FDR<0.05进行负向筛选,lfc>2且FDR<0.05进行正向筛选。
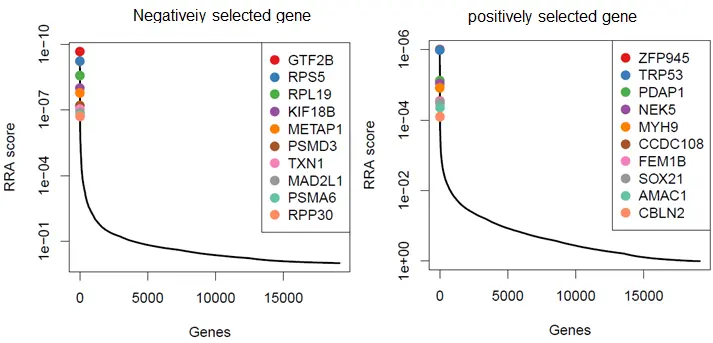
6.GSEA富集分析
对CRISPR筛选得到的候选基因进行富集分析以了解基因的更多功能,同时也可以对CRISPR筛选结果进行验证,以便对潜在候选基因开展进一步研究。
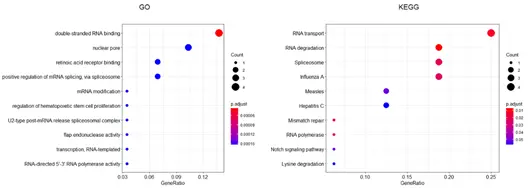
GSEA enrichment analysis
参考文献:
[1]刘燕飞. 基于CRISPR/Cas9技术的HeLa细胞全基因组敲除文库的建立及初步应用[D].中国农业科学院,2020.
[2]Shalem,Ophir, et al. “Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells.”Science, vol. 343, no. 6166, 2014, pp. 84–87.
[3]Zhou, Yuexin, et al. “High-Throughput Screening of a CRISPR/Cas9 Library for Functional Genomics in Human Cells.” Nature, vol. 509, no. 7501, 2014, pp. 487–491.
[4]Kweon, Jiyeon, and Yongsub Kim. “High-Throughput Genetic Screens Using CRISPR-Cas9 System.” Archives of Pharmacal Research, vol. 41, no. 9, 2018, pp. 875–884.
[5]Li, Wei, et al. “Quality Control, Modeling, and Visualization of CRISPR Screens with MAGeCK-VISPR.” Genome Biology, vol. 16, no. 1, 2015, pp. 281–281.
[6]Wang, Binbin, et al. “Integrative Analysis of Pooled CRISPR Genetic Screens Using MAGeCKFlute.” Nature Protocols, vol. 14, no. 3, 2019, pp. 756–780.
作者:打工仔小刘
链接:https://www.jianshu.com/p/6fb8b940e72d
来源:简书
著作权归作者所有。商业转载请联系作者获得授权,非商业转载请注明出处。
想请教一下mageck的mle和rra算法是分别针对了positive selection和nagetive selection嘛?