- 寻找 CDS 序列
NCBI、Ensembl数据库均可以,本人更喜欢Ensembl:http://www.ensembl.org/index.html?redirect=no
2. 分析酶切位点:找出载体多克隆位点 MCS 有,而插入基因中无的酶切位点。
原因:如果序列中也有,酶切会把目的基因CDS序列切开
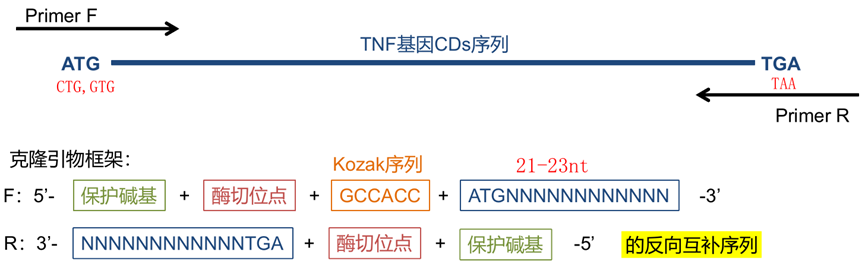
3. 添加保护碱基和 Kozak 序列
Kozak 序列(GCCACC AUG G)是真核生物 5’帽子结构后面的一段核酸序列,可以与翻译起始因子结合,介导含有 5’帽子结构的 mRNA 的翻译起始。对应原核生物的 SD 序列。核糖体可以识别这段序列,并且作为翻译起始位点。
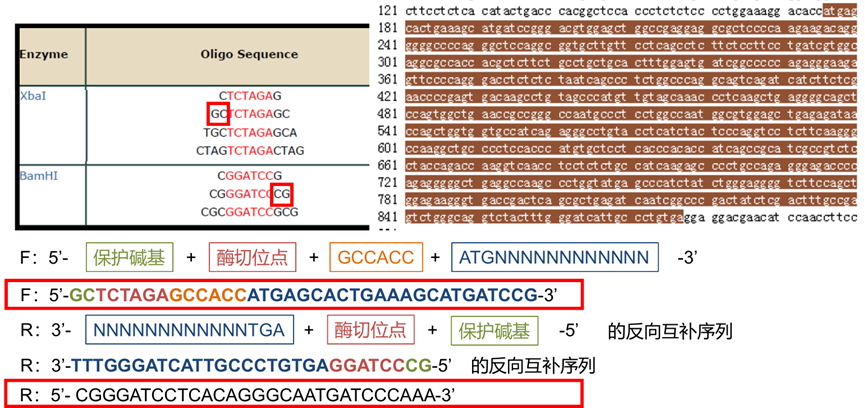
4. 验证引物是否适合 PCR: (不适合也没办法,载体引物的局限性),只看与目的基因互补配对的部分(目的基因 CDS 序列),引物 Tm 值也只计算与目的基因完全互补配对的部分。
5. PCR—>回收—>酶切—>连接—>转化—>筛选—>摇菌—>抽质粒
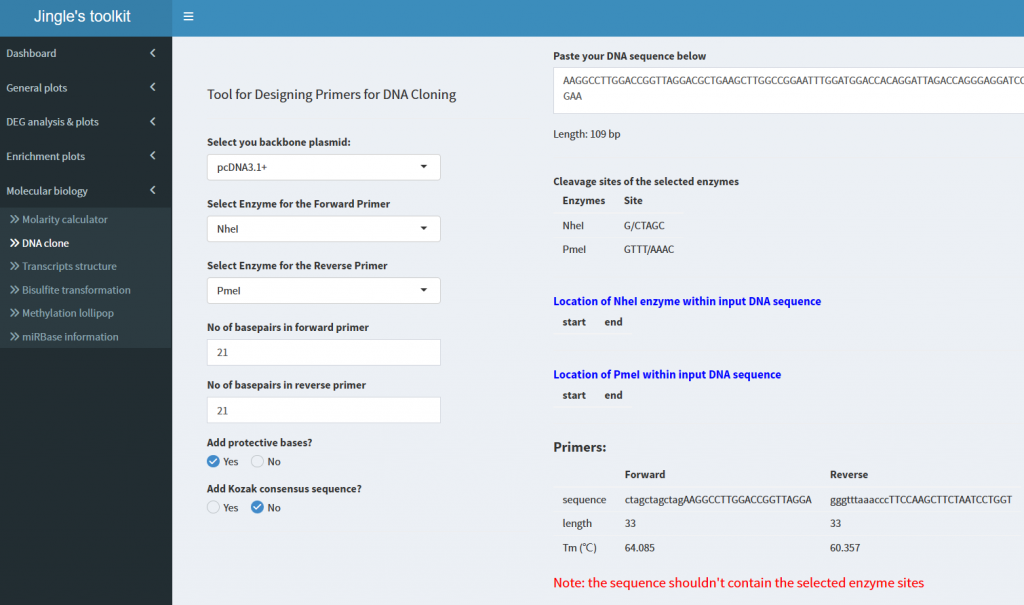
基因克隆引物设计小程序已整合到进哥的toolkit中,有疑问请留言:
您好,请问我想过表达某基因,打算用的载体在C端有EGFP,那么我目的基因与EGFP之间没有T2A或P2A之类的序列,这样表达出来的蛋白是不是就是目的基因与EGFP连在一起的蛋白呢?还有就是,在目的基因与EGFP之间加个2A肽以切割比较好,还是直接两个启动子分别启动它们俩比较好呢?有什么区别吗?不好意思,问题有点多,还望您帮忙解惑!感谢!!!
太感谢啦!好详细的教学步骤。
老师您好,请问酶切位点的保护碱基只需要加5’一端吗?另一端的不需要加吗?(看到其他视频加了两端,所以想在这里请教一下)
酶切位点的保护碱基只需要加在F、R引物的5‘端即可。因为用F、R引物PCR后的到的是双链产物
视频中如果没有5‘端和3’端,过表达结果可能会异常过高或者过低,因为缺乏调控。请教一下,正常一般范围是多少。我用质粒过表达后,PCR结果提示上调大约5倍,这个属于过低吗?
实验室有人用病毒过表达,他PCR结果是上调了50倍。
还有想请教一下,瞬转之后进行PCR检测,使用的引物,就用正常目的基因的引物就可以吧。
谢谢!
你好,RNA水平5倍确实不高,但是应该也够了,过低可能是转染效率过低所致,也就是只有少数细胞转进去了。如果有荧光可以看一下效率。病毒一般效率更高,所以上升倍数更高是正常的,
您好,shRNA和过表达的对照序列,应该怎么设计呢
这个简单,找一个无关序列
哈哈 其实就是人没有的序列,比如GFP绿色荧光蛋白,前提质粒上没有这个,其它任何人没有的都可以
进哥,下游引物的酶切位点和保护碱基需要反向互补么?
哈哈 你可以试一下 反向互补和原序列一样的,常规酶切位点序列是回文序列。
简单点说,不用你做任何变化,直接加5‘端即可’
你好!问一下,1.原核表达正向引物有必要加Kozak序列吗?
2.NCBI上根据目的基因(900bp)设计引物,PCR扩增时发现产物片段短了200bp,可能是什么原因?谢谢!
你好,1.原核不用加的,这是真核表达需要的
2. 可以先查一下目的基因有没有可变剪切产生的不同转录本,可能扩出的其它转录本,实在不清楚可以考虑测序先,PCR产物直接测 第二天就知道结果了