TCGA数据库是一个包括33种癌的各个组学的数据库。我们通过TCGA数据库可以观察每个人的基因表达的变化;甲基化的变化;拷贝数的变化;以及他们的临床信息。
DNA甲基化 (DNA methylation) 是一类发生在胞嘧啶5号位碳原子上、可稳定遗传的表观修饰,在动、植物基因组中广泛存在。不同组织和细胞内的DNA甲基化的建立和模式 (DNA methylation patterns) 对基因调控、转座子沉默及基因印记至关重要。长时间以来,基因组上每个位点上以及不同组织或细胞内的DNA甲基化模式是如何被调控的,是表观遗传学的一个亟待回答的基本问题。
MEXPRESS(https://mexpress.be/)是一个可视化TCGA数据库当中患者的临床信息—甲基化—表达之间之间关系的数据库。进哥以前做甲基化分析一直很依赖这个工具,但是最近,突然这个网站打不开了,也不知道后续会不会恢复。无奈之下,只能另寻他法。众所周知,一个基因上有多个甲基化探针,所以甲基化数据及其大,为了分析单个基因下载全部甲基化数据太亏了。因此,进哥选择从xena browser上下载单个基因甲基化数据,再利用R语言绘图分析。
第一步:数据下载
https://xenabrowser.net/ 点击Visualization
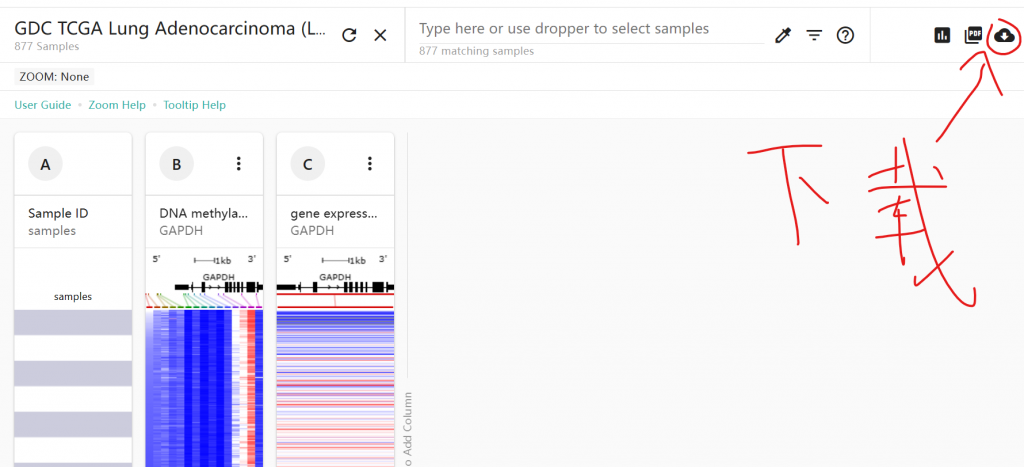
另一个文件是450k甲基化芯片的注释信息,包含每个甲基化探针的位置等信息,可以从GEO或xena下载:illuminaMethyl450_hg38_GDC.txt
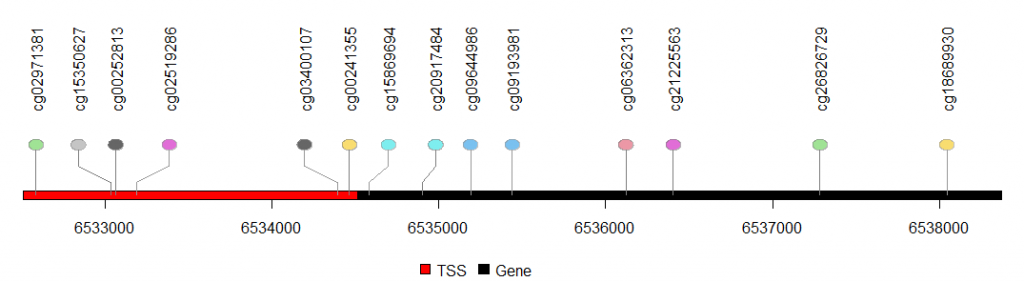
第二步:可视化基因组位置(启动子TSS区和基因body内)
library(trackViewer) #主要画图包
methy <- read.delim("denseDataOnlyDownload.tsv") ##读取Xena下载的数据
anno <- read.delim("illuminaMethyl450_hg38_GDC.txt") ##读取甲基化芯片注释信息
ddd <- anno %>% plotly::filter(X.id %in% colnames(methy))
SNP <- ddd$chromStart
sample.gr <- GRanges("chr17", IRanges(ddd$chromStart, width=1, names=ddd$X.id)) # 设置棒棒图位置
library(biomaRt)
entrez=c("GAPDH")
mart <- useEnsembl(biomart = "ensembl",
dataset = "hsapiens_gene_ensembl")
goids = getBM(attributes = c('hgnc_symbol', 'chromosome_name',"start_position","end_position","strand"),
filters = 'hgnc_symbol',
values = entrez,
mart = mart)
if (goids$strand== -1){
features <- GRanges("chr17", IRanges(c(goids$start_position,goids$end_position+1), # 设置block起使位置
width=c(goids$end_position-goids$start_position, 2000), # 设置block 的长度
names=c("Gene","TSS"))) # 设置名字
features$fill <- c("black","red") #块的颜色
}else{
features <- GRanges("chr17", IRanges(c(goids$start_position-2001,goids$start_position), # 设置block起使位置
width=c(2000, goids$end_position-goids$start_position), # 设置block 的长度
names=c("TSS","Gene"))) # 设置名字
features$fill <- c("red","black") #块的颜色
}
sample.gr$color <- sample.int(length(SNP), length(SNP)) #棒子上面的球的颜色
sample.gr$border <- sample(c("grey60", "grey50"), length(SNP), replace=TRUE) #棒子的颜色
sample.gr$alpha <- 0.6 #设置透明度0-1之间,sample是生成100-200之间的随机数
sample.gr$cex <- 0.5
# sample.gr$label <- "C" #球内的字符
# sample.gr$label.col <- "black" #球内的标签的颜色
# features$height <- c(0.02, 0.05, 0.04) #块的高度
# sample.gr$score <- sample.int(4, length(sample.gr), replace = TRUE) #设置球的数量
lolliplot(sample.gr, features,yaxis = F,ylab = F) #yaxis设置不显示y轴
第三步:相关性分析
results <- psych::corr.test(methy$ENSG00000111640.13,methy[3:(ncol(methy)-1)])
results <- rbind(data.frame(results[["r"]]),data.frame(results[["p"]])) %>% t() %>% as.data.frame()
colnames(results) <- c("cor","p")
r.cut <- 0.3
results$color <- ifelse(results$cor> r.cut,"red",ifelse(results$cor < -r.cut,"green","black"))
results$label <- ifelse(results$p > 0.05,"",ifelse(results$p >0.01,"*",ifelse(results$p>0.001,"**","***")))
sample.gr <- GRanges("chr17", IRanges(ddd$chromStart, width=1, names= paste(ddd$X.id,results[ddd$X.id,]$label))) # 设置棒棒图位置
sample.gr$border <- sample(c("grey60", "grey50"), length(SNP), replace=TRUE) #棒子的颜色
sample.gr$alpha <- 0.6 #设置透明度0-1之间,sample是生成100-200之间的随机数
sample.gr$cex <- 0.5
sample.gr$color <- results[ddd$X.id,]$color #棒子上面的球的颜色
sample.gr$label.col <- "black" #球内的标签的颜色
# features$height <- c(0.02, 0.05, 0.04) #块的高度
# sample.gr$score <- sample.int(4, length(sample.gr), replace = TRUE) #设置球的数量
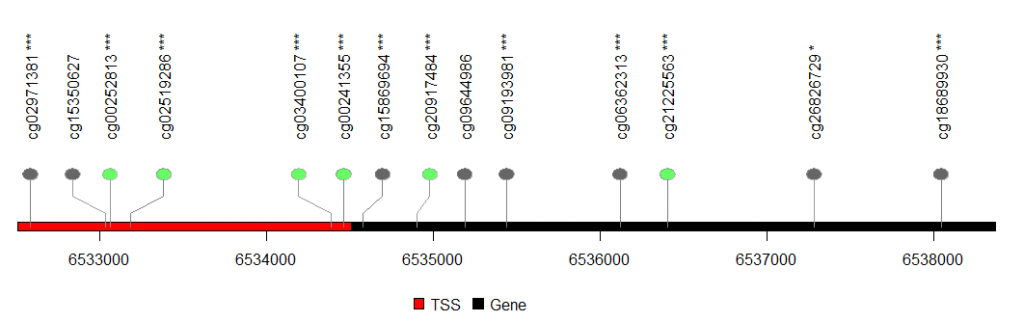
lolliplot(sample.gr, features,yaxis = F,ylab = F) #yaxis设置不显示y轴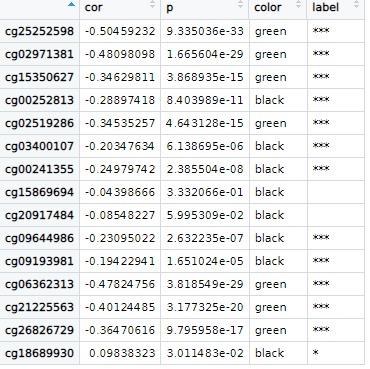
你好请问goids结果不止一行是什么原因呢
进哥你好,为什么我选的那个基因,画出来的图上面没有结果?
进哥,为什么我在results里面做出了表达和甲基化的关系,也有星号。但是最后一步绘图却出现了以下报错:
Warning message:
In .Seqinfo.mergexy(x, y) :
The 2 combined objects have no sequence levels in common. (Use
suppressWarnings() to suppress this warning.)
进哥,df2是data数据啊,出现这种错误请问是为什么呢
Error in count_N[count_N$n < 4, ]$Tissue :
$ operator is invalid for atomic vectors
chr17这个位置是通用的吗?别的基因也用这个位置?不是咋改?谢谢
进哥你好,我想请教一下,我看一篇文章做了单基因的甲基化和表达量的相关性分析,但是图中只有一个cor值,请问这里可以直接取所有位点甲基化的平均值,然后再与表达量做相关性分析吗。原文作者并没有讲述如何处理的。谢谢!
其实我也不清楚这一个值哪来的,也许有什么算法进行了计算吧。。。
一直报错
Error in GRanges(“chr1”, IRanges(ddd$chromStart, width = 1, names = ddd$X.id)) :
could not find function “GRanges”
是怎么回事呀,纯小白
已经加微信了是吧 这个问题就是 没有加载相应的包
‘ranges’ must have the length of the object to construct (1) or length 1,你好,请问出现这种情况应该怎么办啊?
你是加我微信的那位同学吗?检查一下你的数据里面有没有甲基化和表达数据
是的是的,已经解决了,谢谢
第三步:相关性分析中间有一步代码错误,会导致星号错位,已更正:
sample.gr <- GRanges("chr17", IRanges(ddd$chromStart, width=1, names= paste(ddd$X.id,results[ddd$X.id,]$label))) # 设置棒棒图位置
请问老师这里的chr17是否需要改为自己结果中对应的染色体,例如您B站视频中的结果为chr12,这步里是否需要改为chr12呢?
非常好的教程!!!就是对于我这种临床学生来讲,需要去了解一些甲基化和基因转录的一些基础知识,继续努力,加油!
B站来的!!感谢大哥!
不客气,有需要一起交流
感谢进哥,马上去对着入手