应B站粉丝留言,咱们出一个GEO多数据集联合分析的教程,以及shiny app,分享代码这么就,总是还有人搞不定,索性代码和app一起奉上。
众所周知,高通量数据平台非常之多,大类咱们分为芯片数据和测序数据。而细分而言,芯片平台又有很多,每个平台都有自己的注释文件,而相关信息可能不一定一致。比如对于我们研究最主要的基因名字(通常我们都是用的Official gene symbol),由于基因命名前后经过很多次更新,因此几年前的芯片注释文件中的基因名字可能与现在的有所差异,而这种差异就会给我们的多数据及整合分析带来一定的麻烦,可能遗漏某些重要基因。
因此,在进行多数据集整合分析时建议将基因名称转化为official gene symbol,这个其实比较简单,limma包提供了这样一个函数:alias2symbol,使用方法如下:
alias2Symbol(alias, species ="Hs", expand.symbols =FALSE)
alias2SymbolTable(alias, species ="Hs")
考虑到上述这个原因,我还是建议大家在提取GEO数据时可以使用Gene ID、Ensemble ID这些比较稳定的基因Identifier进行分析,最后再将其转化成Symbol。关于ID转化的教程,我B站有分享,大家网上也能查到不少方法,这不赘述。
在这个小程序中。针对基因名字标准化,进哥设置了三个模块,分别对应几种情况的处理:
①Symbol extract:注释文件没有给出单独的基因名称或ID,而是如下一串:NM_005562 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918 /// NM_018891 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918 /// XM_006711316 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918 /// ENST00000264144 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918 /// ENST00000493293 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918 /// BC112286 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918 /// BC113378 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918 /// AK296944 // LAMC2 // laminin, gamma 2 // 1q25-q31 // 3918。处理原则就是提取genebank accession,也就是NM_…,然后进行ID转换成gene symbol。
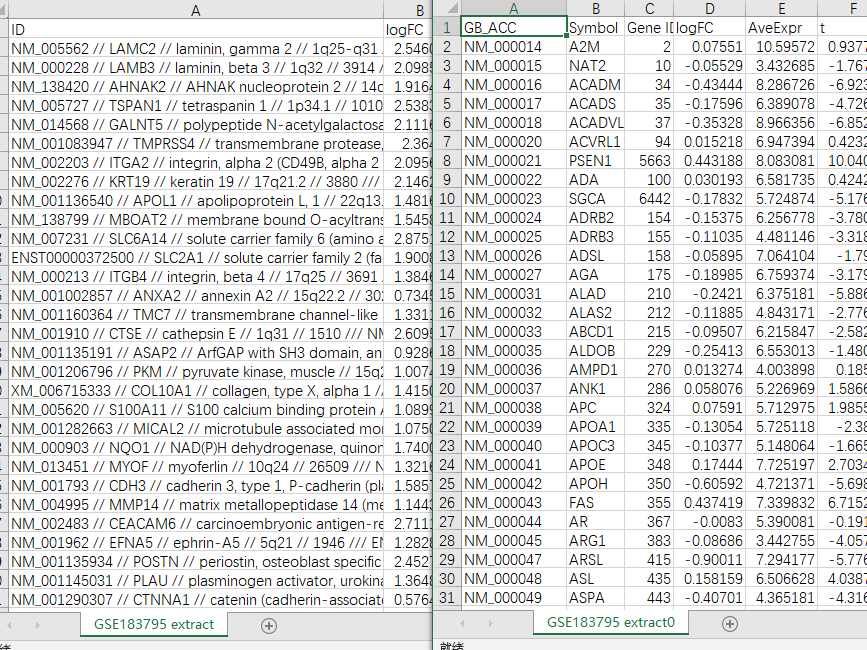
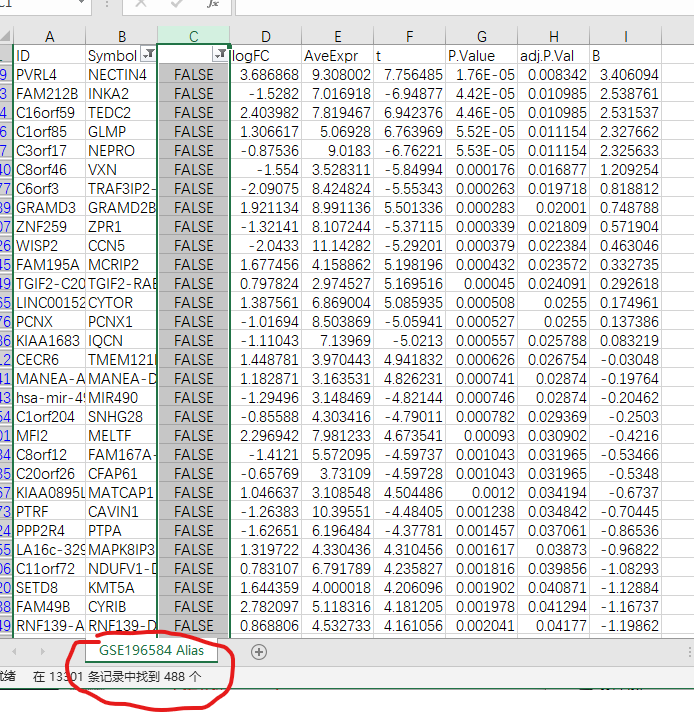
②Alias to Symbol:别名转换,这个案例中有488个official gene symbol与gpl注释文件中不一致。
③ID convert:包括Gene ID/Ensembl ID/GB_Acc转换成Symbol
接下来进入正题:就是多数据集联合分析,提供了三种方法:
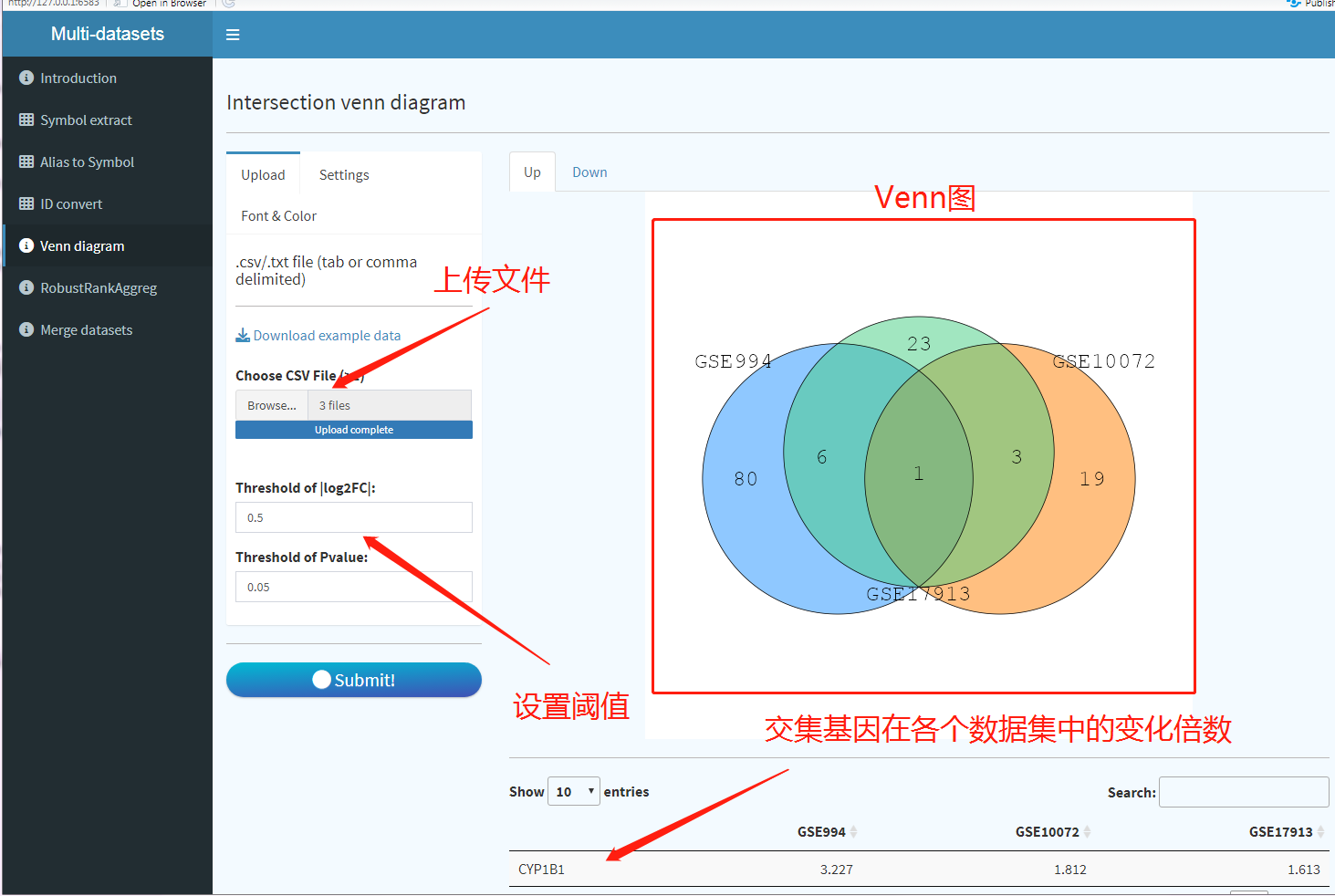
①Venn取交集:上传基因symbol标准化之后的limma包差异分析结果,直接多选需要分析的DEG分析结果文件(仅支持CSV和txt),不能修改limma分析结果中的列名。对于显著基因筛选阈值,可以自己设置;
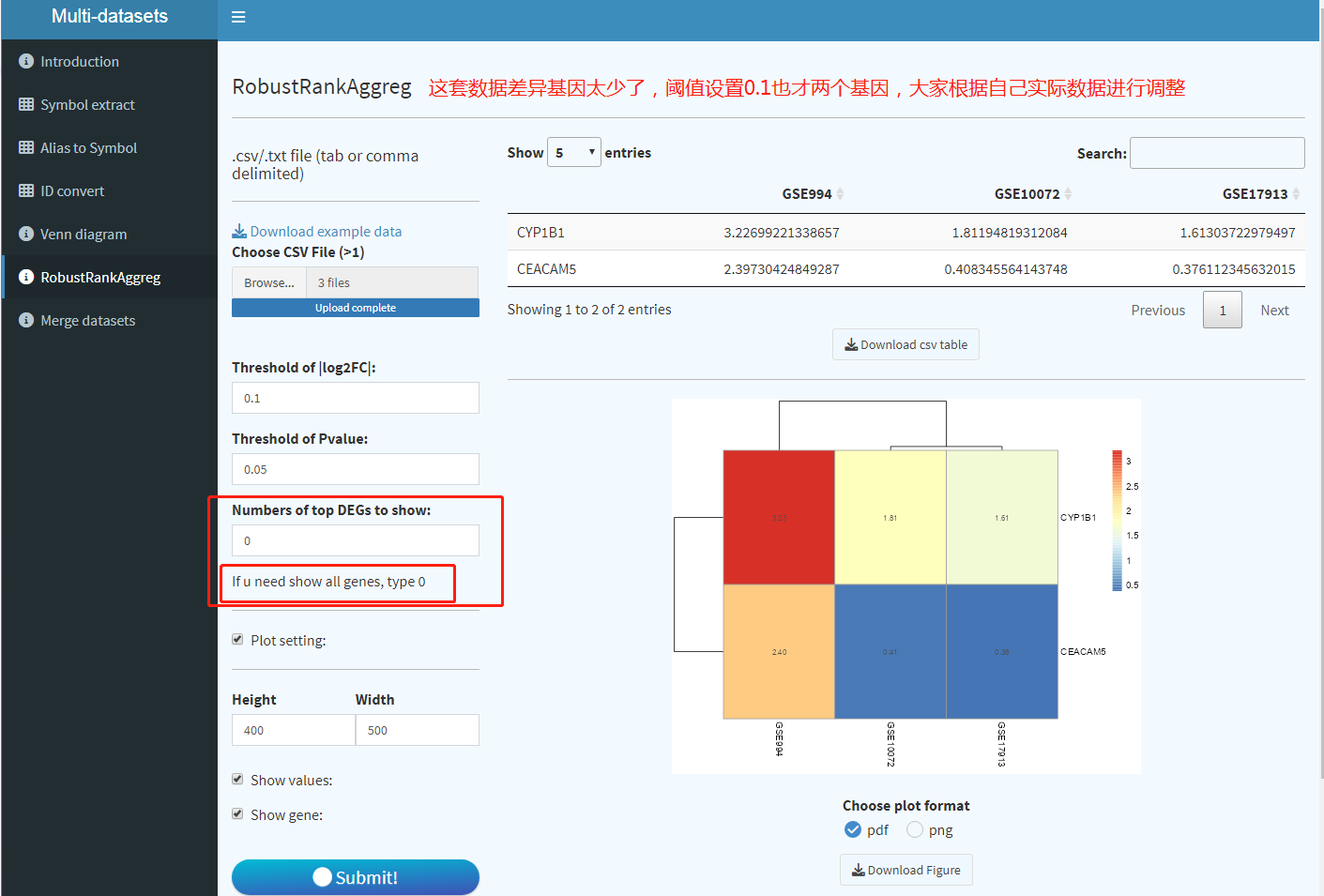
②RobustRankAggreg
RRA(Robust Rank Aggregation)是一种对排名进行整合,获得一个综合性排名列表的算法。这个方法最早实在2012年发表在Bioinformatics杂志上。
在数据挖掘过程中,我们经常可能会遇到这种情况。例如我们同时分析了三套表达谱数据,都得到了差异表达分析结果。
那么我们手上会有3个差异表达的gene list。怎么样才能挑出一些更重要的或者更有生物学意义的基因做后续分析或者实验验证呢?我们通常的做法可能是直接取这三个gene list的交集。虽然这样做确实可行,但是这种方法只考虑到了gene出现的次数,而没有考虑到基因在三个list里面的排序。
RRA方法其实就是对多个排好序的基因集,进行求交集的同时还考虑一下它们的排序情况。总体上来说,就是挑选那些在多个数据集都表现差异的基因,并且每次差异都排名靠前的那些,他们的最终的综合排名也会比较靠前。
模块界面与Venn相似,同样上传差异分析结果文件,设置阈值以及Top基因的数目。如果我们需要全部排名的基因,将number of top DEGs to show设置为0.
③多数据集整合,去批次化:基于Combat
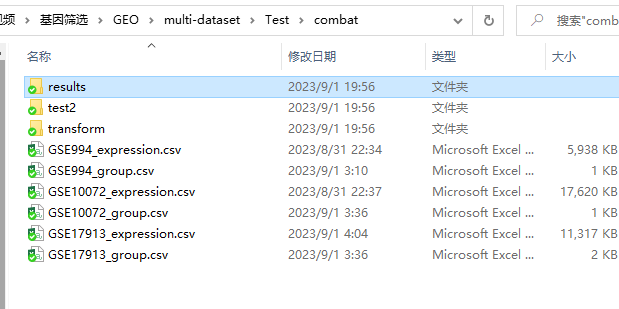
将各个数据集的分组文件和数据文件存放到一个目录下,注意,不管是该脚本还是app,表达矩阵格式应该为:数据集名称_expression.csv,而分组文件应该为:数据集名称_group.csv,如下图。当然除了csv,txt也支持。
下面是分析主体代码:
library(sva)
library(dplyr)
##定义数据所在目录,随后操作由程序自动运行
datapath <- '.../multi-dataset/Test/combat'
group <- c("Never","Current")
list_files <- list.files(datapath,pattern = "csv|CSV|txt|TXT")
filetype <- strsplit(list_files[1],"[.]")[[1]][2]
fff <- strsplit(list_files,"_")
datasets <- lapply(1:length(fff), function(x){
fff[[x]][1]
}) %>% unlist() %>% unique()
if (filetype %in% c("csv","CSV")){
group_data <- read.csv(paste0(datapath,"/",datasets[1],"_group.",filetype))
expr_data <- read.csv(paste0(datapath,"/",datasets[1],"_expression.",filetype))
}else{
group_data <- read.delim(paste0(datapath,"/",datasets[1],"_group.",filetype))
expr_data <- read.delim(paste0(datapath,"/",datasets[1],"_expression.",filetype))
}
if (max(expr_data[-1]) > 100){
expr_data[,2:ncol(expr_data)] <- log2(expr_data[,2:ncol(expr_data)])
}
group_data$group <- factor(group_data$group, levels = group)
group_data <- group_data[order(group_data$group),]
group_data$batch <- 1
expr_data <- cbind(expr_data[1], expr_data[-1][group_data[,1]])
colnames(expr_data)[1] <- "DATA"
for (ddd in datasets[-1]) {
if (filetype %in% c("csv","CSV")){
gg <- read.csv(paste0(datapath,"/",ddd,"_group.",filetype))
dd <- read.csv(paste0(datapath,"/",ddd,"_expression.",filetype))
}else{
gg <- read.delim(paste0(datapath,"/",ddd,"_group.",filetype))
dd <- read.delim(paste0(datapath,"/",ddd,"_expression.",filetype))
}
if (max(dd[,-1]) > 100){
dd[,2:ncol(dd)] <- log2(dd[,2:ncol(dd)])
}
colnames(dd)[1] <- "DATA"
gg$group <- factor(gg$group, levels = group)
gg <- gg[order(gg$group),]
gg$batch <- which(datasets == ddd)
dd <- cbind(dd[1], dd[-1][gg[,1]])
group_data <- rbind(group_data,gg)
expr_data <- merge(expr_data,dd,by = "DATA")
}
expr_data <- tibble::column_to_rownames(expr_data,var = "DATA")
expr_data[is.na(expr_data)] <- min(expr_data, na.rm=TRUE)/2
expr_data <- expr_data[which(!apply(expr_data,1,sd)==0),]
group_data$batch <- as.numeric(group_data$batch)
library("sva")
library(limma)
mod <- model.matrix(~as.factor(group),data = group_data )
expr_batch <- ComBat(dat = expr_data,
batch = group_data$batch,
mod = mod,
par.prior = T) ##sd=0至此就得到整合的矩阵,后续进行limma包差异分析:
# 构建对比矩阵
design <- model.matrix(~ 0 + group_data$group)
colnames(design) <- levels(factor(group_data$group))
rownames(design) <- group_data$Accession
fomula <- paste0(group[2],"-",group[1])
contrast.matrix<-makeContrasts(contrasts= fomula,
levels = design)
contrast.matrix##查看比较矩阵的信息,这里我们设置的是Treat Vs. control
fit <- lmFit(expr_batch,design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit2 <- eBayes(fit2)
DEG<-topTable(fit2, coef=1, n=Inf) %>% na.omit() ## coef比较分组 n基因数
DEG<-tibble::rownames_to_column(DEG,var = "Gene")
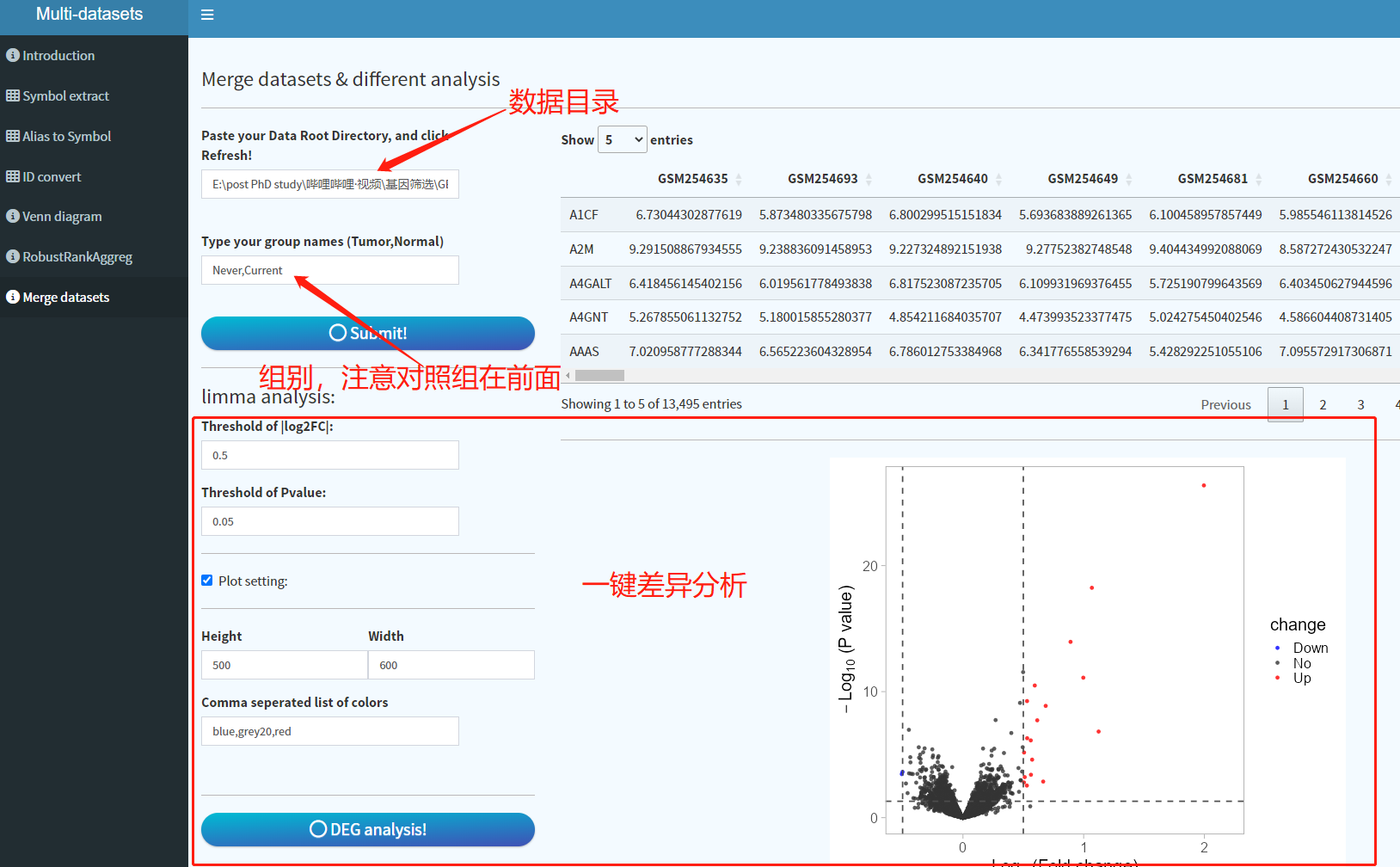
App就是完全基于该脚本,并多出了可视化结果 也就是火山图。这个不复杂,也许我的代码和别人在数据处理上稍有不同,但是最终目的是一样,我这样的目的是为了让尽量多的工作让R语言执行,而非自己整理,这当然有需要一点基础,不然万一有一点数据上的问题,就无法进行了。下面是小程序该模块截图:
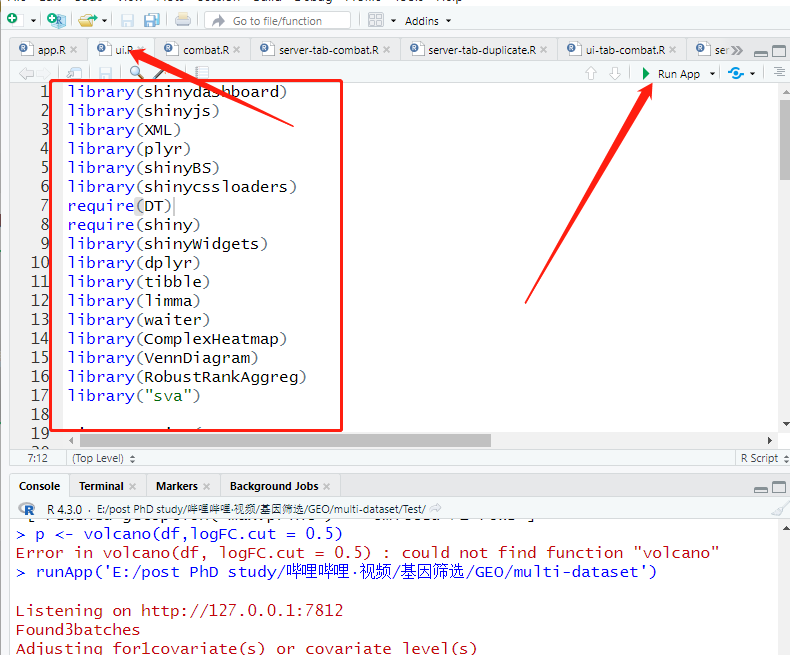
最后讲一下APP的打开方式,你需要的R基础就是安装包,打开ui.R文件,下图红框中的包需要安装好,然后点击右上角Run App即可,若提示没有安装那个包,回到R界面命令窗口安装好即可(主要install.packages & BiocManager::install两种方式)。
你好 为什么运行不了
你好,怎么运行不了啊