肿瘤不是单纯的恶性细胞群,而是由不同类型细胞组成的复杂生态系统。在这些细胞中,肿瘤浸润免疫细胞在肿瘤控制和治疗反应中起着核心作用。例如,细胞毒性CD8 + T细胞是抗癌免疫的主要效应因子,因为它们可以特异性地识别和杀死携带新抗原的肿瘤细胞。肿瘤特异性抗原主要来源于突变基因的表达。但免疫细胞也可以发挥免疫抑制作用,支持肿瘤发生和免疫逃避,如调节性T细胞。因此,对不同类型的肿瘤浸润免疫细胞进行定量研究,有助于阐明肿瘤免疫应答的机制、评价肿瘤治疗的免疫原性作用,最终指导设计合理治疗方案。背景介绍最著名的标记基因分析方法是基因集富集分析(GSEA)。基于GSEA的方法计算富集分数(ES),当某一细胞类型的特异性基因在感兴趣的样本中处于高表达的前几位,而在其他情况下则处于低表达的前几位时,该分数较高。
基于GSEA的方法只能计算样本中细胞类型富集的半定量分数,而反褶积方法可以定量地估计感兴趣的细胞类型的相对分数。反褶积算法将异种样本的基因表达谱看作是不同细胞的基因表达水平的卷积,并利用描述细胞类型特异性表达谱的特征矩阵来估计未知的细胞组分。
下面是利用标记基因与GSEA或其他评分方法,或利用反褶积算法和免疫细胞表达特征,从细胞混合物的表达数据量化免疫细胞的计算方法。

下图是几个从转录组学数据定量肿瘤浸润免疫细胞的计算工具:
M = marker genes, P = partial deconvolution, C = complete deconvolution
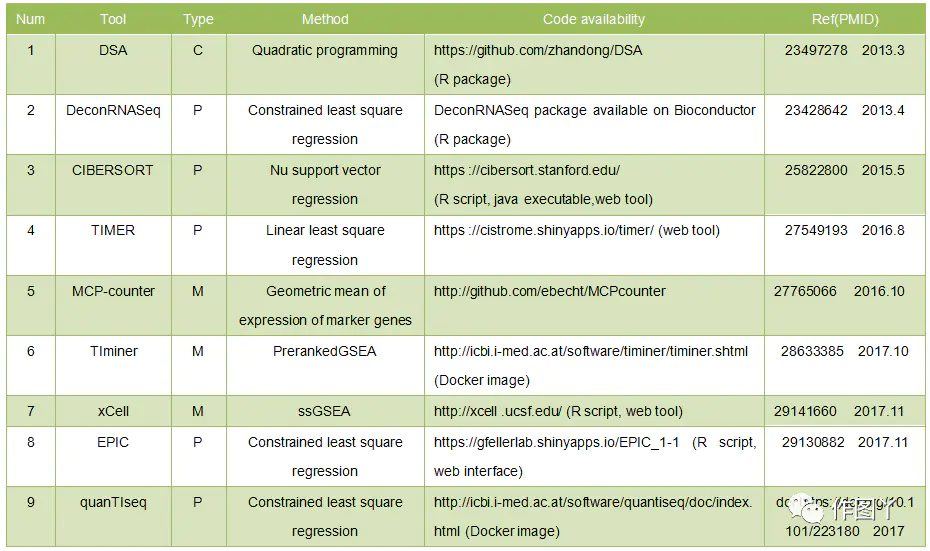
方法简介****01基于标记基因的评分方法(1)MCPcounter
一种基于标记基因定量肿瘤浸润免疫细胞(CD3 + T细胞,CD8 + T细胞,细胞毒性淋巴细胞,NK细胞,B淋巴细胞,单核细胞来源的细胞(单核细胞系),髓样树突状细胞,中性粒细胞)、成纤维细胞和上皮细胞的方法。对于每个细胞类型和样本,丰度得分为细胞类型特异性基因表达值的几何平均值,独立地对每个样本进行计算的。由于分数是用任意单位表示的,它们不能直接解释为细胞分数,也不能在细胞类型之间进行比较。进行定量验证时,估计分数与真实细胞分数之间具有很高的相关性,证明了MCP-counter用于样本间比较的价值。MCP-counter已应用于对32个非血液学肿瘤的19,000多个样本中的免疫细胞和非免疫细胞进行量化。
(2)TIminer
TIminer是一个用户友好的计算框架,用于不同的肿瘤免疫基因组分析,包括(i)来自NGS数据的人白细胞抗原HLAs基因分型;(ii)使用突变数据和HLA类型预测肿瘤新抗原;(iii)来自大量RNA-seq数据来分析定量肿瘤浸润免疫细胞;(iv)通过表达数据量化肿瘤的免疫原性。
(3)xCell
xCell是一种基于ssGSEA的方法,能够计算64种免疫细胞的丰度分数,包括适应性和先天免疫细胞、造血祖细胞、上皮细胞和细胞外基质细胞。基于FANTOM5,ENCODE,Blueprint,Immune Response In Silico (IRIS),Human Primary Cell Atlas (HPCA)和Novershtern等6个研究的489个基因集。对于每种细胞类型,计算xCell丰度分数的主要步骤有四个:(i)利用R包GSVA对489个基因集单独进行ssGSEA;(ii)对属于一种细胞类型的所有基因集的ES进行平均;(iii)平台特异性ES转化为丰度分数;(iv)使用与流式细胞术数据分析类似的spillover方法纠正密切相关的细胞类型之间的关系。
02使用表达特征对细胞混合物进行反褶积(1)DeconRNASeq
Gong和Szustakowski将约束回归模型应用于RNA-seq数据分析,并将其实现在R包DeconRNASeq中。混合五种人体组织(大脑、骨骼肌、肺、肝和心脏)的RNA-seq数据,通过合并组织类型特异性基因来估计组织或细胞类型的比例。并利用Illumina’s human Body Map 2.0中RNA-seq数据构建的特征矩阵,对算法进行了仿真验证。虽然还没有开发出新的免疫特征,但该工具原则上可以应用于任何特征矩阵。
(2)CIBERSORT
CIBERSORT算法利用微阵列数据构建特征矩阵,描述22种免疫细胞表型的表达特征,包括不同的细胞类型和功能状态的免疫细胞。CIBERSORT使用ν-SVR评估细胞分数。CIBERSORT分别在9个免疫细胞亚群和3个免疫细胞亚群的同时反褶积方面具有较高的准确性,在4种恶性免疫细胞的模拟混合物上测试,也证明了对不同程度的噪音和未知的肿瘤具有鲁棒性。
(3)TIMER
一个系统评估不同的免疫细胞对不同癌症类型的临床影响的资源。利用一系列免疫特异性标记和免疫细胞表达特征,来估计32种癌症类型中6种免疫细胞类型(B细胞、CD4 T细胞、CD8 T细胞、中性粒细胞、巨噬细胞和树突状细胞)的丰度。从RNA-seq或微阵列数据中提取的癌症表达矩阵与免疫细胞表达矩阵合并,并用Combat进行归一化,以消除批量效应。通过从免疫细胞标记中选择与肿瘤纯度负相关的基因,为每种癌症类型分别识别特征基因。最后,对于每种癌症类型,考虑到所选的免疫细胞标记,从标准化的免疫细胞配置文件构建特征矩阵。TIMER使用线性最小二乘回归方法执行反褶积,并强制所有负估计为零。用越来越小的一组T细胞标记重复估计几次,以减少CD8 +和CD4 + T细胞比例之间的相关性。与CIBERSORT不同的是,最终的估计并没有被规范化,因此不能直接解释为细胞组分,也不能在不同的免疫细胞类型和数据集之间进行比较。
(4)EPIC
Estimate the Proportion of Immune and Cancer cells (EPIC),用来估计免疫细胞和癌细胞比例。EPIC使用约束最小二乘回归明确地将非负性约束引入反褶积问题,并要求每个样本中所有细胞组分的总和不超过一个。EPIC克服了以往从大量肿瘤基因表达数据预测癌症和免疫细胞或其他非恶性细胞类型的方法的几个局限性,考虑了非特征性和可能高度可变的细胞类型,并在算法上得到了发展。能够广泛应用于大多数实体肿瘤,如黑色素瘤和结直肠样本验证的情况,但不适合血液恶性肿瘤,如白血病或淋巴瘤。
(5)quanTIseq
quanTIseq是专门为RNA-seq数据开发的反褶积工具,能够准确定量未知肿瘤的含量,以及量化整体组织的免疫细胞组份。基于约束最小二乘回归和一个新的特征矩阵(来自51个纯化或富集的免疫细胞类型的RNA-seq数据集)。quanTIseq实现了一个完整的反褶积流程来分析RNA-seq数据,能够避免混合物和特征矩阵之间的不一致性。
03细胞组分和表达谱的同时反褶积(1)DSA
数字排序算法Digital Sorting Algorithm—DSA是一个完整的反褶积算法,它基于一组从混合组织样本中提取在特定细胞类型中高度表达的标记基因,利用二次规划来推断复杂组织中的细胞组分和表达谱。该算法在三种恶性免疫细胞系混合的微阵列数据上进行了测试,重建了真实的细胞组分和表达谱。该算法是无偏的,不需要细胞类型频率的先验知识。
REF:Finotello, F., & Trajanoski, Z. (2018). Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer immunology, immunotherapy : CII, 67(7), 1031–1040. https://doi.org/10.1007/s00262-018-2150-z
您好,我想问一下,蛋白组学可以做免疫浸润吗,这种处理合理吗
wow, 哈哈哈哈发现了宝藏主页,同样28岁公卫博士在读,免疫浸润学习了,感谢分享
哈哈 谢谢认可 一起学习