老大在群里出的题,说感觉这个热图很诡异,然后中间我自己没有用
boxplot查看数据的表达量,对于数据不能有正确的认识,导致一开始的deg的logFC都没有达到正负1的,最重要的是:1.是因为我知道什么情况下用
log函数,然后我还在这里面错误的用了log函数;2.不能用
[1:4,1:4]查看数据集机构;3.
normalizeBetweenArrays和removeBatchEffect函数的用处。老大帮助修改了代码,关于
normalizeBetweenArrays、removeBatchEffect和boxplot的,结果才清晰明了。
原文图片
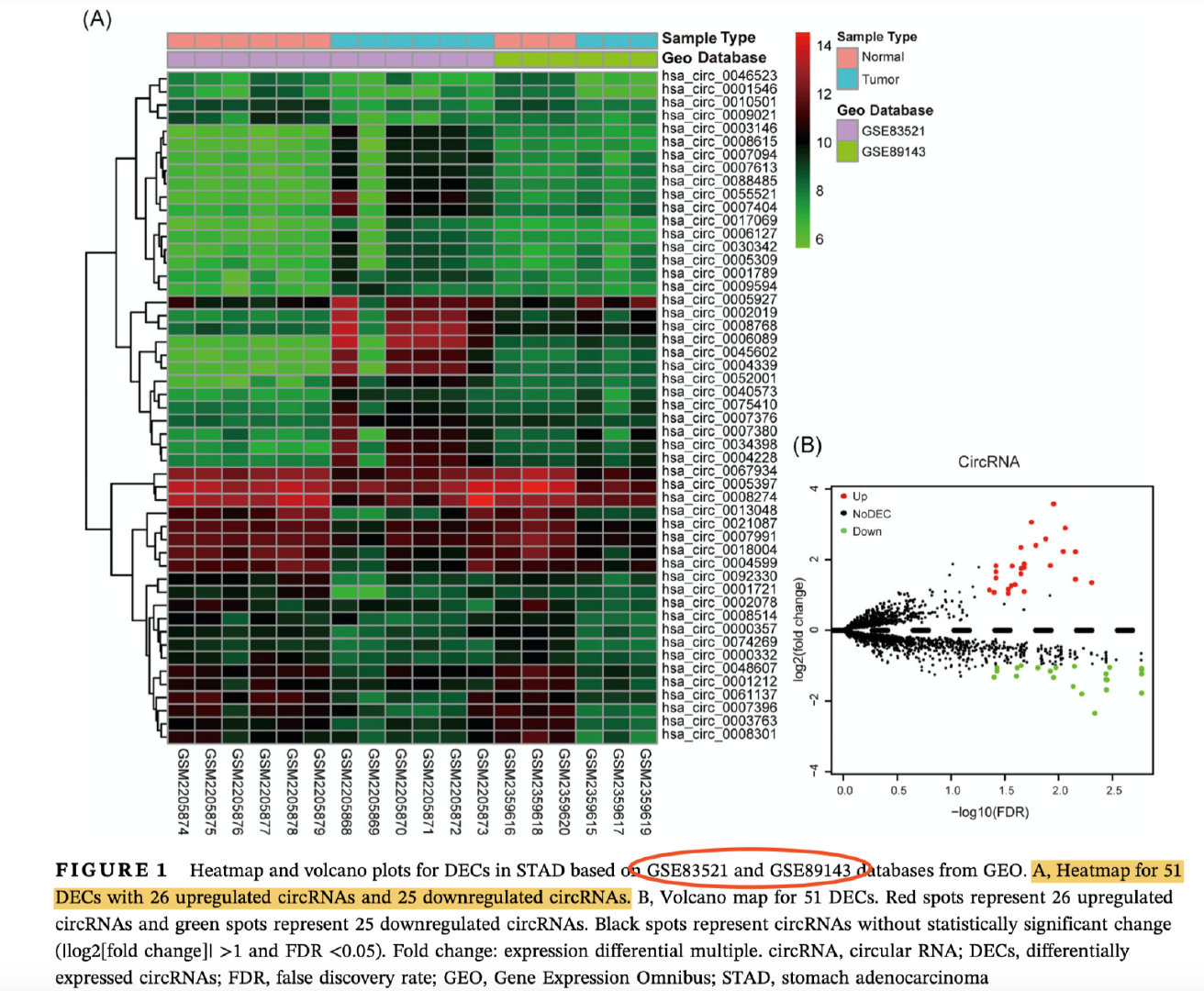
下载数据+准备数据
rm(list = ls())
options(stringsAsFactors = F)
library(GEOquery)
eSet1 <- getGEO("GSE83521",
destdir = '.',
getGPL = F)
#(1)提取表达矩阵exp
exp1 <- exprs(eSet1[[1]])
exp1[1:4,1:4]
dim(exp1)
# 一定要看boxplot
boxplot(exp1,las=2)
pd1 <- pData(eSet1[[1]])
#(3)提取芯片平台编号
gpl1 <- eSet1[[1]]@annotation
save(pd1,exp1,gpl1,file = "step1-1output.Rdata")
load("step1-1output.Rdata")
eSet2 <- getGEO("GSE89143",
destdir = '.',
getGPL = F)
#(1)提取表达矩阵exp
exp2 <- exprs(eSet2[[1]])
exp2[1:4,1:4]
dim(exp2)
boxplot(exp2,las=2)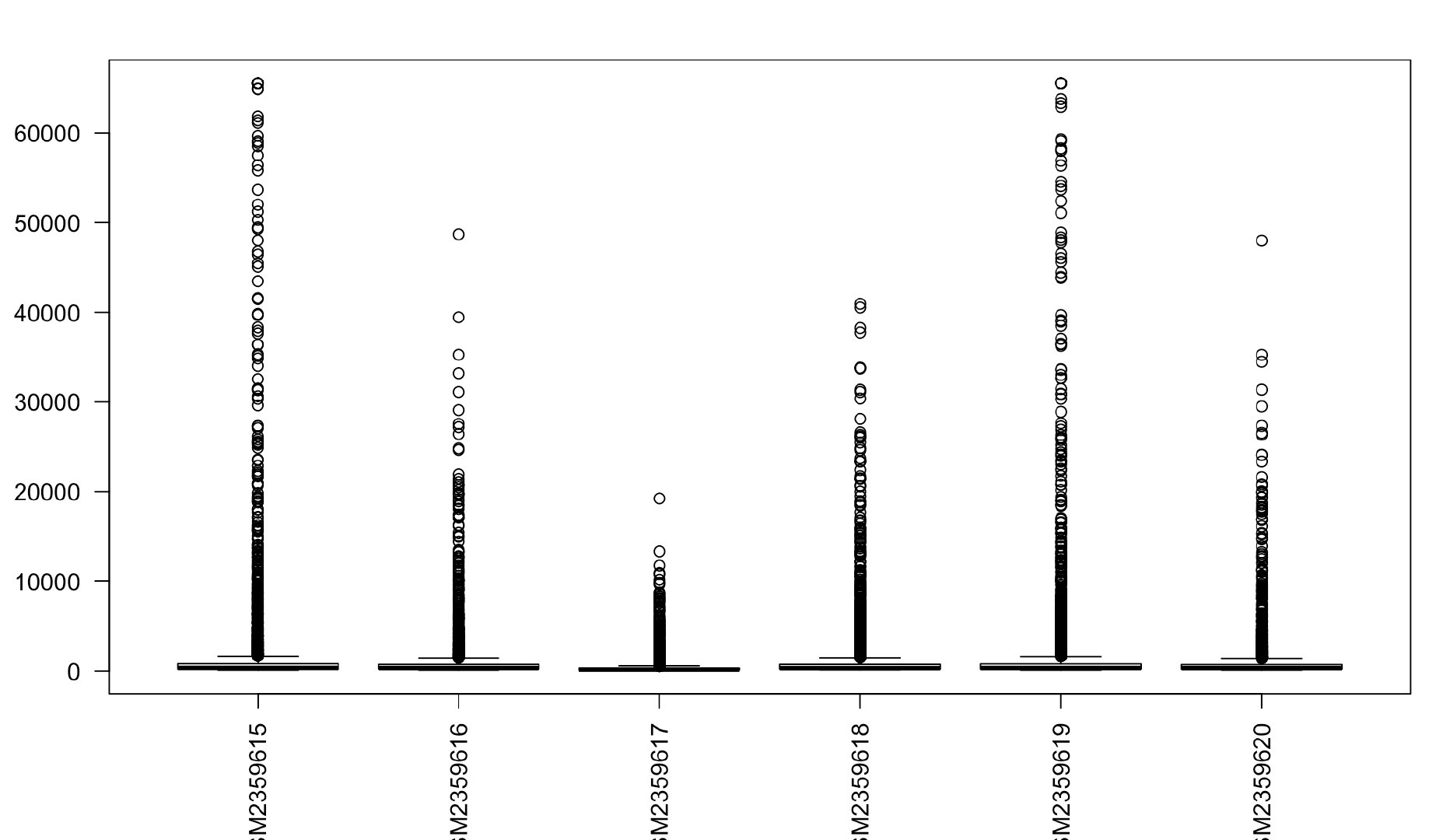
从上面的箱线图结果可以看到,数值的表达量并不在同一条水平线上,并且有成败上千,也有零,很明显是没有经过log的。这是需要把数据log后再用boxplot来看数据的分布,用boxplot来看数据的分布非常重要。不能仅仅用[1:4,1:4]来查看,因为[1:4,1:4]并不能看到整体的数据情况。关于为什么要log,是因为做差异分析的limma包要求表达矩阵中的数据是经过log的。可以参考老大的这篇:关于limma包差异分析结果的logFC解释
exp2 = log2(exp2+1)
boxplot(exp2,las=2)
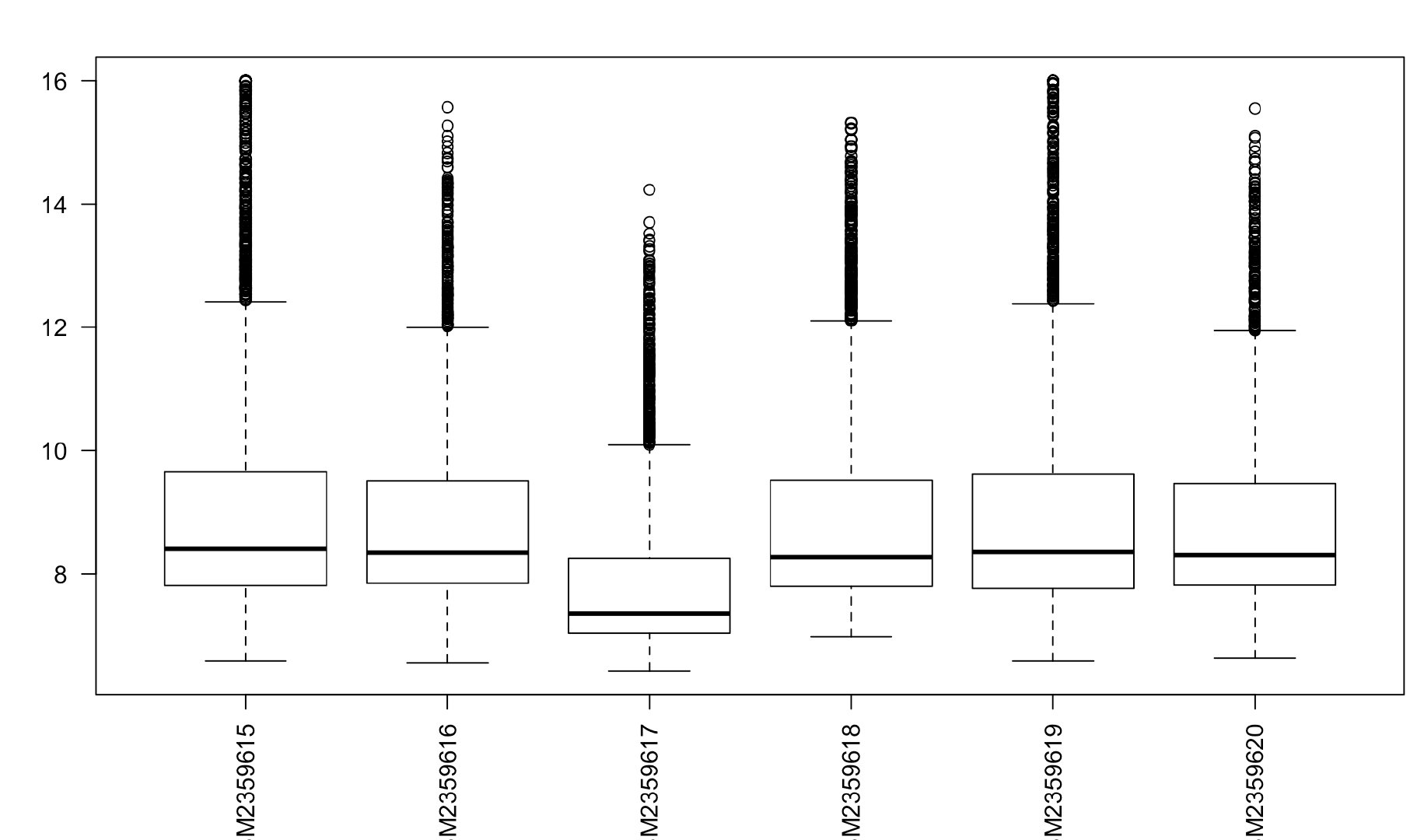
接下来这个函数厉害了,从上面的图中可以看到有一个样本的中位数和其他样本明显不在一条水平显示,这个normalizeBetweenArrays函数,可以把他拉回正常水平,normalizeBetweenArrays只能是在同一个数据集里面使用。
library(limma)
exp2=normalizeBetweenArrays(exp2)
boxplot(exp2,las=2)

从上面的箱线图可以看到,exp2的数据的分布基本在一条水平线上。
接下来将exp2的数据保存
#(2)提取临床信息
pd2 <- pData(eSet2[[1]])
#(3)提取芯片平台编号
gpl2 <- eSet2[[1]]@annotation
#这些代码是什么鬼东西,我给你注释了。
index <- sort.int(pd2$characteristics_ch1,index.return = T)
class(index)
pd2 <- pd2[index$ix,]
exp2 <- exp2[,match(rownames(pd2),colnames(exp2))]
save(pd2,exp2,gpl2,file = "step1-2output.Rdata")
load("step1-2output.Rdata")探针注释
## 是同一个平台,非常棒
gpl2
gpl1
if(T){
library(GEOquery)
gpl<- getGEO('GPL19978', destdir=".")
dim(gpl)
colnames(Table(gpl)) #查一下列明
head(Table(gpl)[,c(1,2)])
ids=Table(gpl)[,c(1,2)]
ids <- ids[-c(1:2),]
ids <- ids[-c(1:13),]
save(ids,file='ids.Rdata')
}看一下获得的探针和circ_RNA的对应关系
差异分析-去除批次效应
x1 <- exp1[rownames(exp1) %in% ids$ID,]
x2 <- exp2[rownames(exp2) %in% ids$ID,]
boxplot(x1,las=2)
boxplot(x2,las=2)
cg=intersect(rownames(x1),rownames(x2))
x_merge=cbind(x1[cg,],x2[cg,])- 又有大招了,得到的这个
merge后的表达矩阵x_merge,一定一定要用boxplot看一下,因为我们是将两个数据集通过共同的探针合并了,因为是来自两个数据,所以第一次在实际案例中接触到了这个高大上的名次-去除批次效应。 - 那么就
boxplot来看看!
boxplot(x_merge)
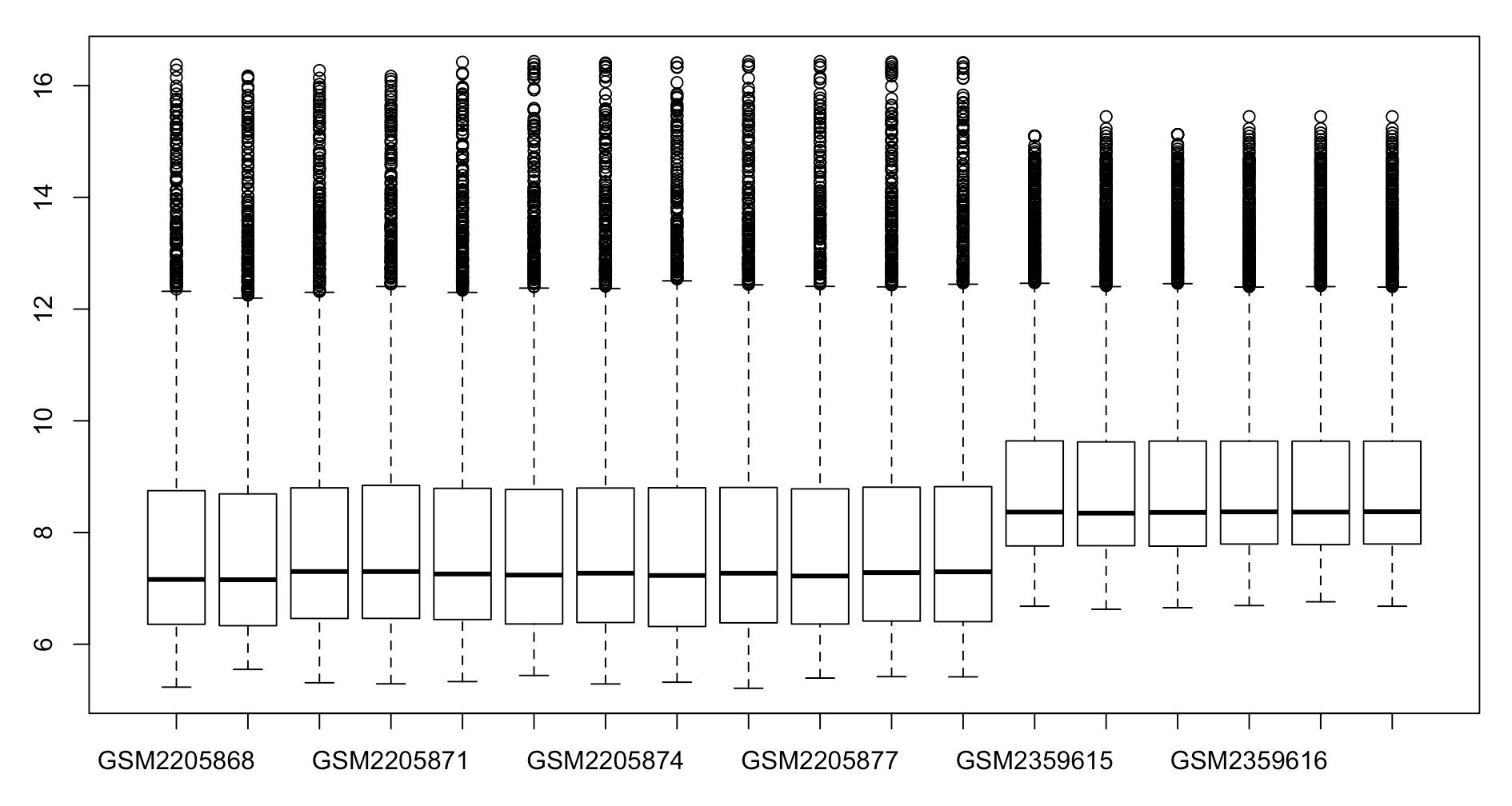
上面这张图,就是非常明显的看到了,由于后面的三个样本就是来自另一个数据集的。从前面的boxplot(exp2)也可以看到他的表达量在8以上。
- 这样就需要去除批次效应,参考多种批次效应去除的方法比较
- 但是还需要做一些数据准备
pd1$title
pd2$characteristics_ch1
group_list <- c(rep('tumor',6),rep('normal',6),rep(c('tumor', 'normal'),each=3))
gse <- c(rep('GSE83527',12),rep('GSE89143',6))
table(group_list,gse)
dat <- x_merge
library(sva)
library(limma)
## 使用 limma 的 removeBatchEffect 函数
dat[1:4,1:4]
batch <- c(rep('GSE83521',12),rep('GSE89143',6))
design=model.matrix(~group_list)
ex_b_limma <- removeBatchEffect(dat,
batch = batch,
design = design)
dim(ex_b_limma) - 这个时候一定要看 boxplot , 不然就白学的了!!!!!
boxplot(ex_b_limma)
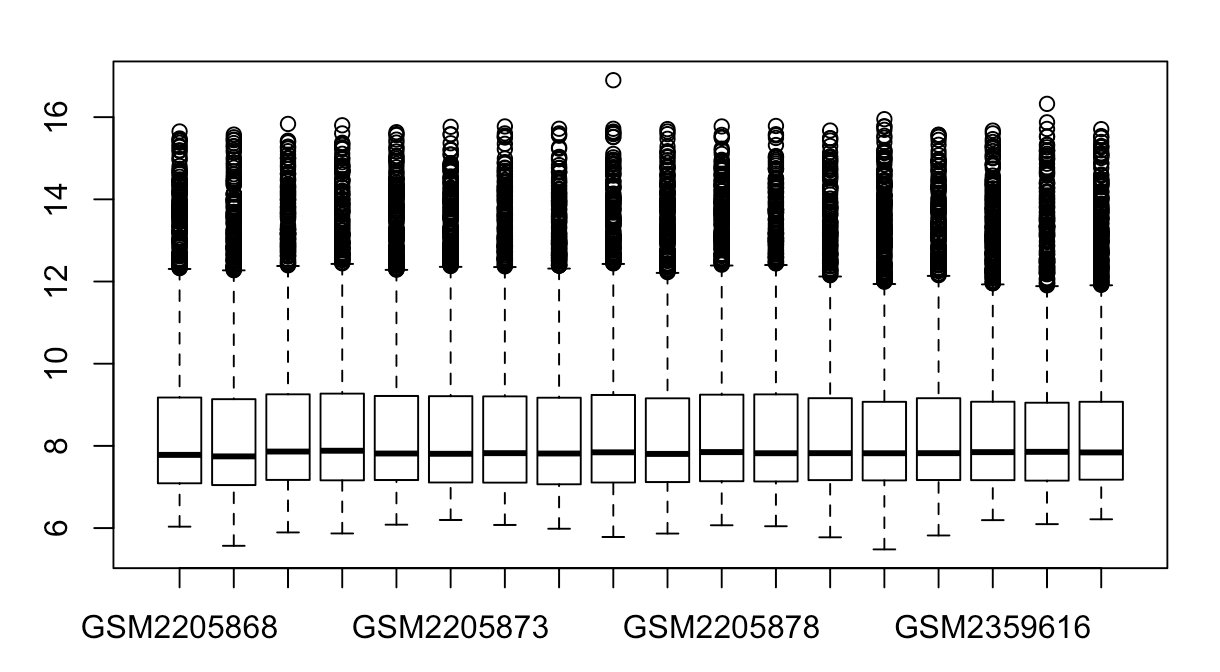
- 从上面的图可以看到了,去除了批次效应后,数据的表达水平。
接下来就做差异分析
{
library(limma)
fit=lmFit(ex_b_limma,design)
fit=eBayes(fit)
options(digits = 4)
topTable(fit,coef=2,adjust='BH')
deg=topTable(fit,coef=2,adjust='BH',number = Inf)
}
得到的deg如下图
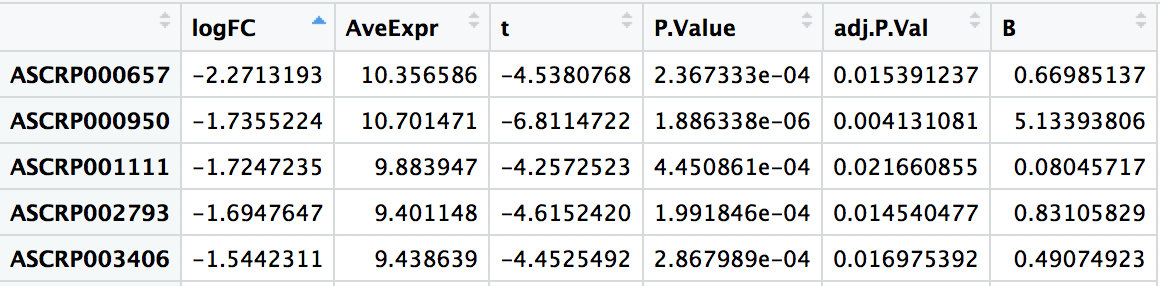
?两张图主要是为了看logFC的值,我第一次把两个数据全部log并且没有进行一个normalizeBetweenArrays来去除样本间的批次差异,而没有一个logFC值是在>1和
火山图
if(T){
nrDEG=deg
head(nrDEG)
attach(nrDEG)
plot(logFC,-log10(P.Value))
library(ggpubr)
df=nrDEG
df$v= -log10(P.Value) #df新增加一列'v',值为-log10(P.Value)
ggscatter(df, x = "logFC", y = "v",size=0.5)
df$g=ifelse(df$P.Value>0.05,'stable', #if 判断:如果这一基因的P.Value>0.01,则为stable基因
ifelse( df$logFC >1,'up', #接上句else 否则:接下来开始判断那些P.Value<0.01的基因,再if 判断:如果logFC >1.5,则为up(上调)基因
ifelse( df$logFC < -1,'down','stable') )#接上句else 否则:接下来开始判断那些logFC <1.5 的基因,再if 判断:如果logFC <1.5,则为down(下调)基因,否则为stable基因
)
table(df$g)
df$name=rownames(df)
head(df)
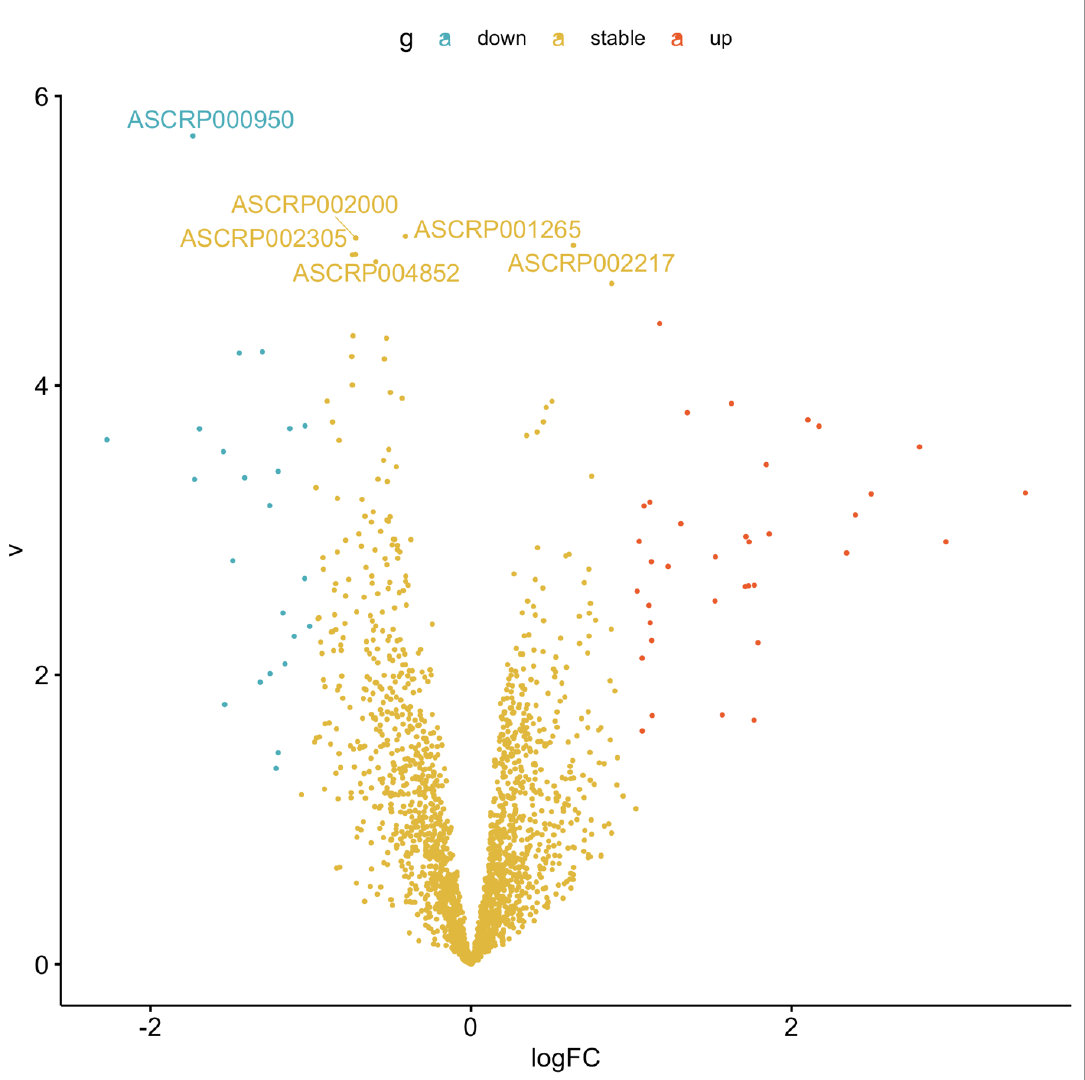
ggscatter(df, x = "logFC", y = "v",size=0.5,color = 'g')
ggscatter(df, x = "logFC", y = "v", color = "g",size = 0.5,
label = "name", repel = T,
#label.select = rownames(df)[df$g != 'stable'] ,
label.select = head(rownames(deg)), #挑选一些基因在图中显示出来
palette = c("#00AFBB", "#E7B800", "#FC4E07") )
ggsave('volcano.png')
} 
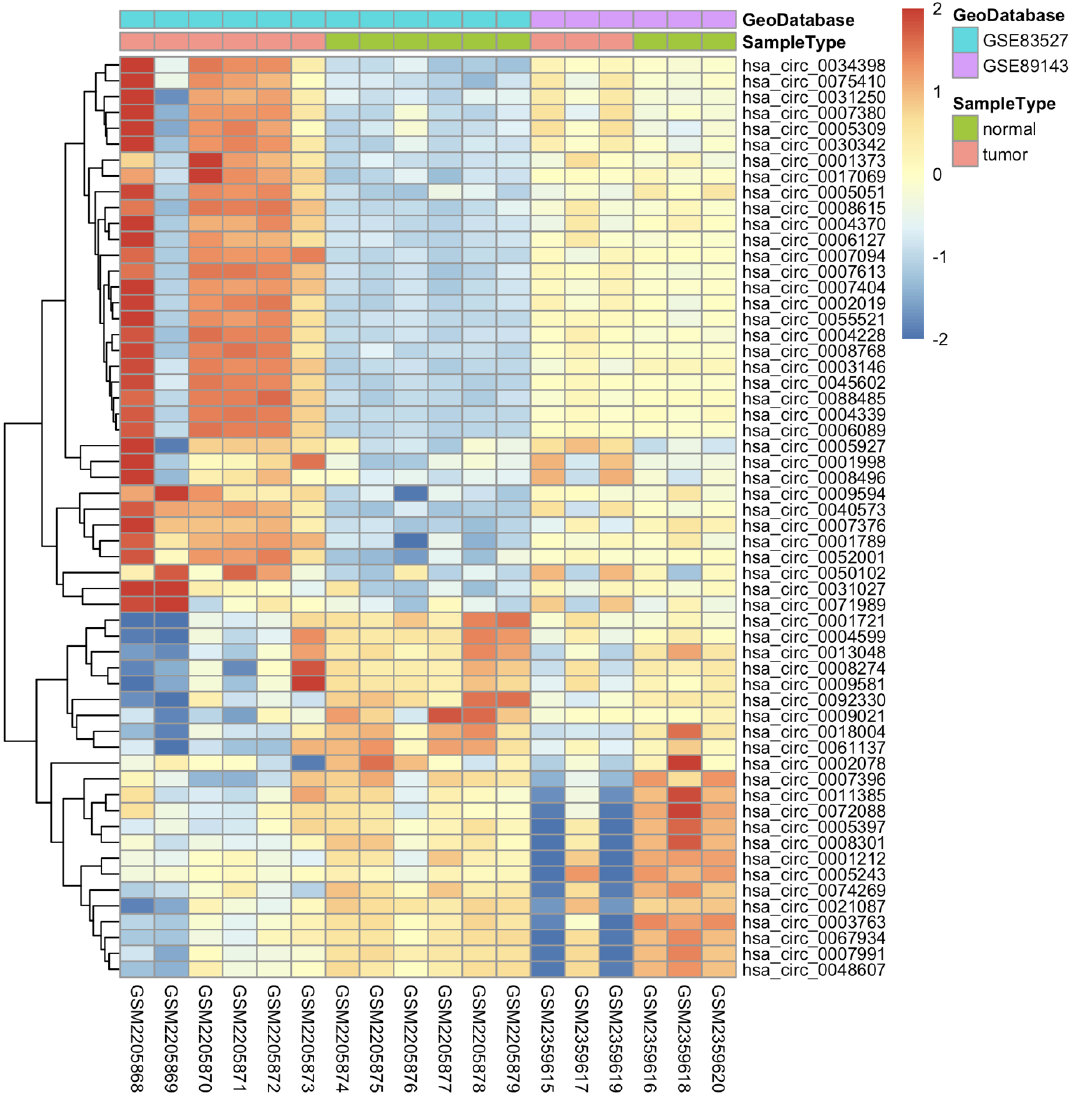
热图
- 在画特图前,还有一小问题,那么就是这个探针的问题,我们可以看到原文的热图上的基因是
hsa-circ-0034398,而我们id转换后的表达矩阵的基因名是,如下图标记所示

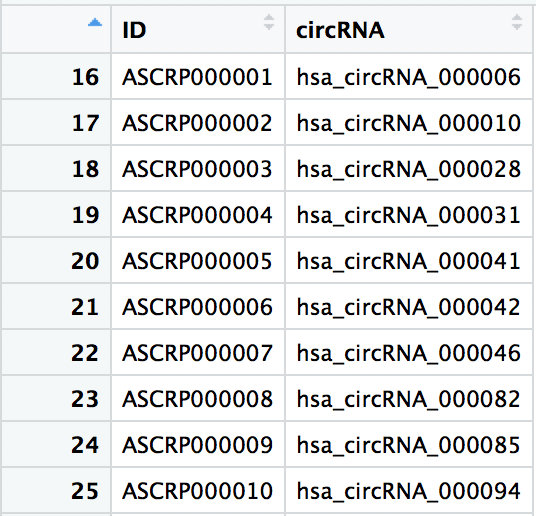
学习群里的小伙伴将一个Alias的对应表格分享到群里,把它读进R里进行进一步的转换,文件是ID.txt。
if(T){
up <- df[df$g=='up',]
down <- df[df$g =='down',]
x <- rbind(up,down)
x$ID <- rownames(x)
#上面是为了提取出差异基因的子集
y <- merge(x,ids,by='ID') #merge函数可以根据两列共有的内容进行合并,合并后含有共有的行名
ex_b_limma2 <- ex_b_limma[match(y$ID,rownames(ex_b_limma)),]
rownames(ex_b_limma2) <- y$circRNA
#上面的函数写的不咋好,命名变量不好命名,需要得到结果后看一下
z <- read.csv('ID.txt',sep = '\t')
tmp1 <- z[z$circRNA %in% rownames(ex_b_limma2),]
ex_b_limma3 <- ex_b_limma2[match(tmp1$circRNA,rownames(ex_b_limma2)),]
rownames(ex_b_limma3) <- tmp1$Alias
library(pheatmap)
pheatmap(ex_b_limma3,show_colnames =T,show_rownames = T)
n=t(scale(t(ex_b_limma3)))
n[n>2]=2
n[n< -2]= -2
n[1:4,1:4]
pheatmap(n,show_colnames =F,show_rownames = F)
ac=data.frame(SampleType=group_list,#这步就是可以实现两个分组了
GeoDatabase=gse)
rownames(ac)=colnames(n) #将ac的行名也就分组信息 给到n的列名,即热图中位于上方的分组信息,这步很重要
pheatmap(n,show_colnames =T,
show_rownames = T,
cluster_cols = F,
annotation_col=ac,
fontsize = 8,
filename = 'deg-heatmap.png') #列名注释信息为ac即分组信息
}
重要的如下:
1.理解
boxplot的重要性,来看数据集是否需要log,以便后面才能用limma包进行差异分析2.
normalizeBetweenArrays只能是在同一个数据集里面用来去除样本的差异,不同数据集需要用limma 的removeBatchEffect函数去除批次效应。
进哥您好,想问一下您GEO数据联合分析的时候能使用高通量测序的和RNASEQ的一起去除批次效应后分析,找差异基因吗?谢谢!
请教大佬,如果这两个数据集不是同一个芯片平台,并且不打算矫正批次效应的情况下,想要将两个数据的箱线图展现在一个图中,直接rbind数据合理嘛?请问有更好的处理方法吗?
你好,rbind肯定行不通,merge或innerjion合并 根据基因列
进哥哥,你好
removeBatchEffect函数的原理大概是抹平两个数据集的变化差异,那如果我想分析两个样本对象不一样的数据集,如一个是患者组,一个是对照组,对这两组进行removeBatchEffect的话不会把患者与对照的差异给去除吗?
如果你的一个数据只有患者,另一个数据只有对照,而且还有不确定的批次效应,那me你的数据应该是用不了的
您好,我想问两份的GEO数据是log2后,但是箱图都不齐的,如果要合并的话。是二者分别标准化再合并去批次?还是log2还原原始数据再合并去批次标准化?又或者是log2不用还原合并去批次再标准化?合并、去批次、标准化三者之间的顺序究竟应该是怎样的?
请教,有两次RNA-seq的数据,如何去除批次效应?
你好,这个我没有研究过,我给您找了一份资料参考,有问题再讨论
去除RNA-seq数据批次效应_qq_27390023的博客-CSDN博客_rna测序批次效应https://blog.csdn.net/qq_27390023/article/details/123306912