STRING网站的官网(网址为https://cn.string-db.org/),左侧一栏是网站的输入方式,右侧一栏是我们的蛋白输入框。通常我们在做PPI时选择输入方式是“Multiple proteins”,即多个蛋白的输入。
1. Input your genelist,并选择对应物种🡪Search
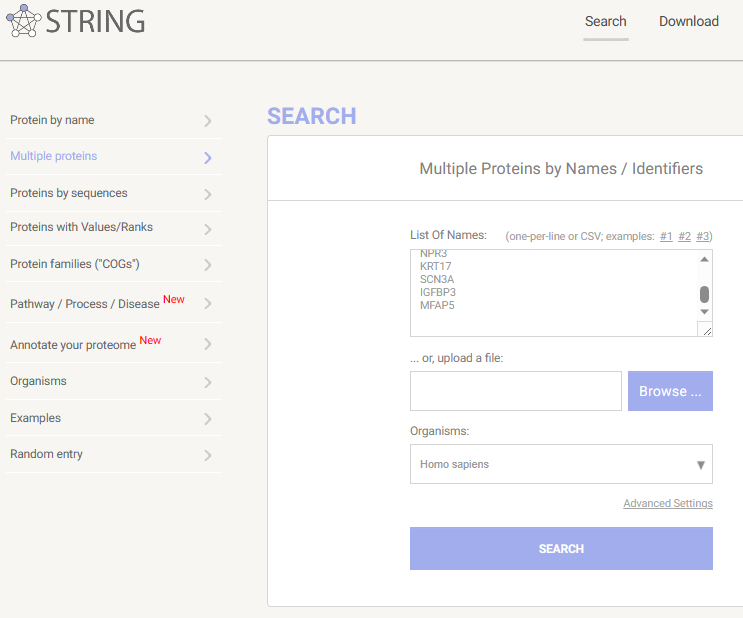
2. Mapping gene symbol
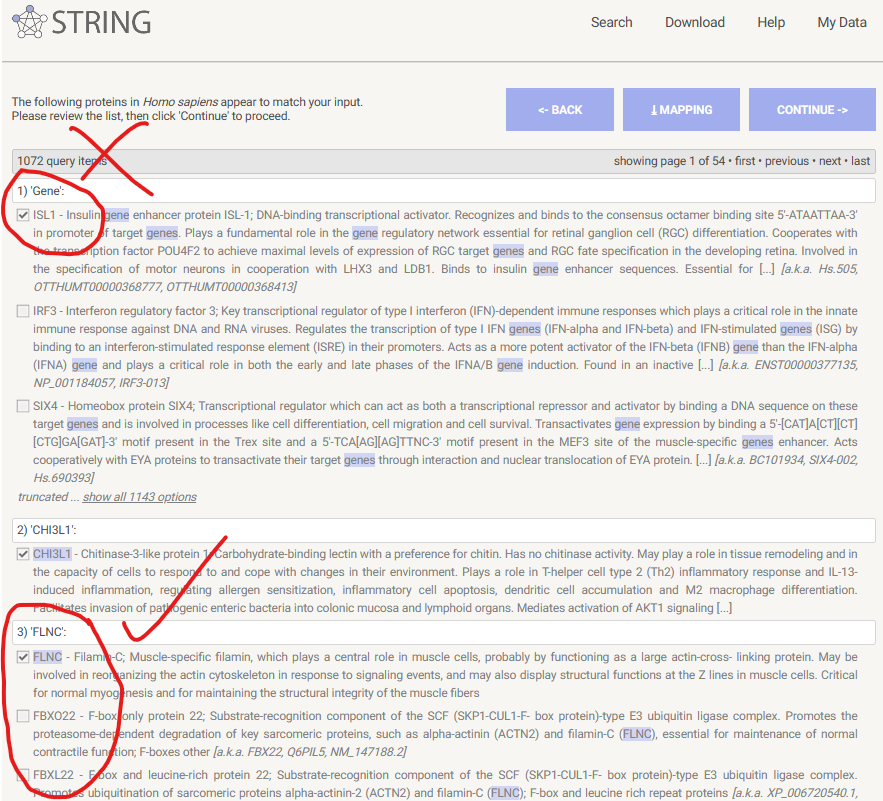
这一步看似平平无奇,实则也很重要,大家看画圈之处应该能看出端倪。
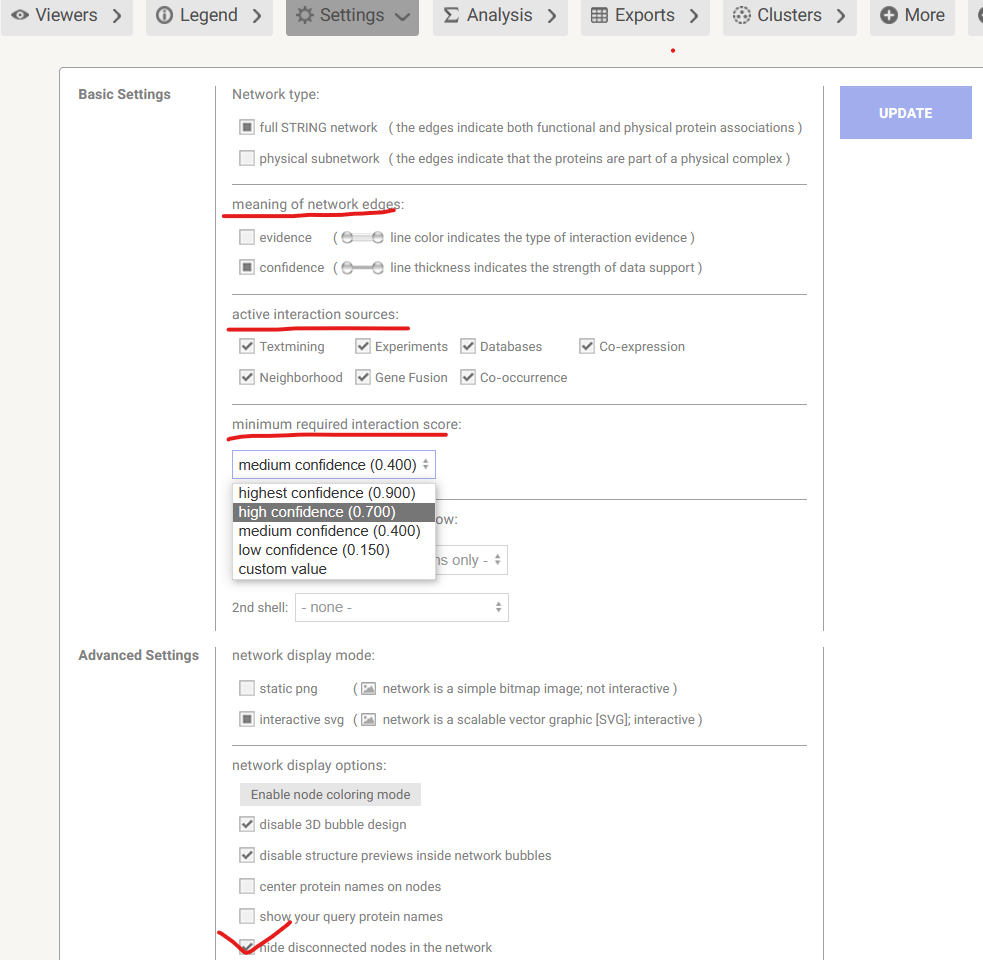
3. PPI network setting
本案例差异基因表较多,所以默认参数下网络过于复杂:

这种情况下我们需要根据实际情况调整参数:
点击setting:
如下图划线部分参数均可调整,已根据要求生成符合自己要求的网络。
我一般主要调整minimum required interaction score为:high confidence或者highest confidence;
此外,勾选hide disconnected nodes in the network,
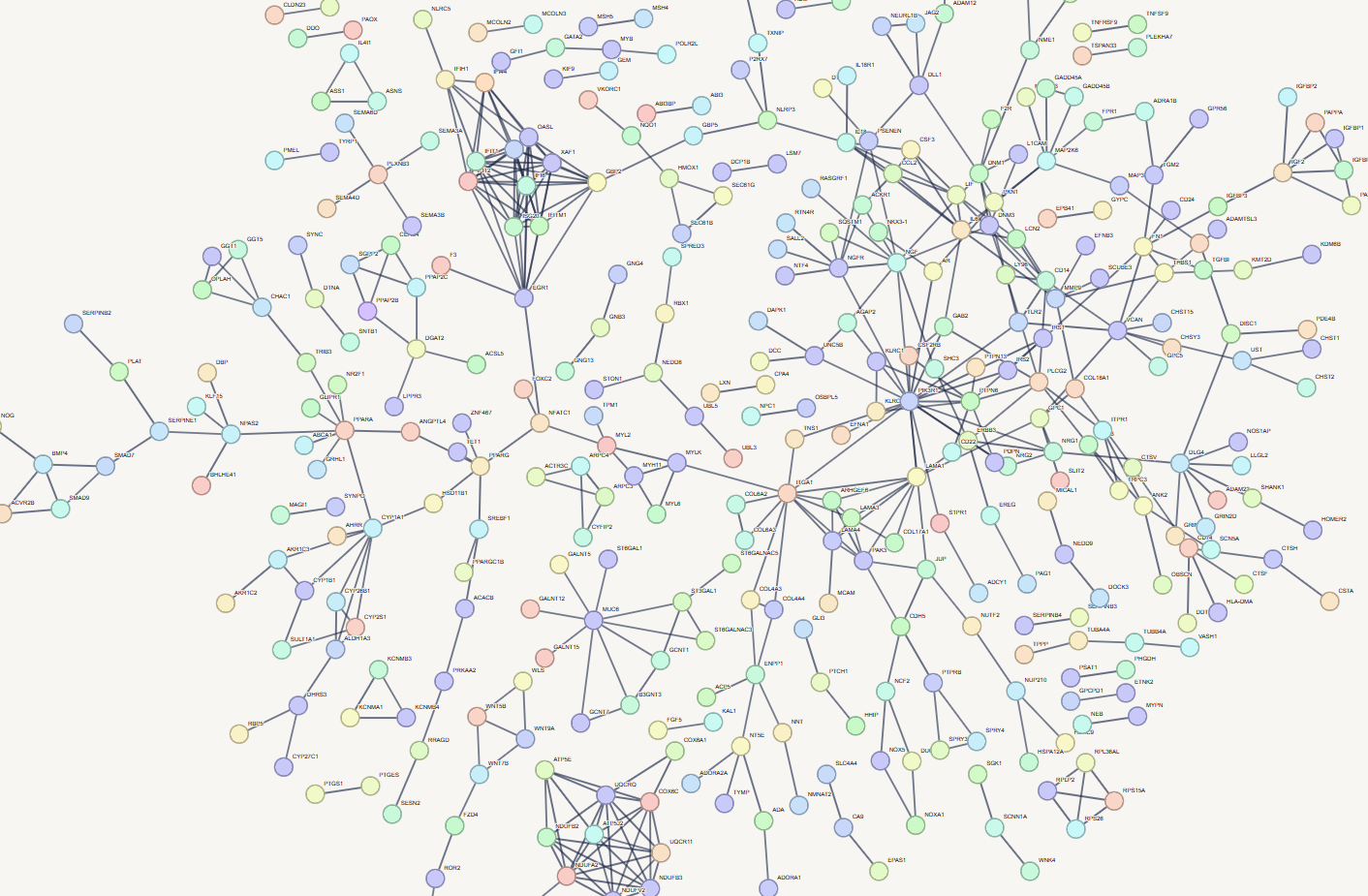
Update(highest confidence):
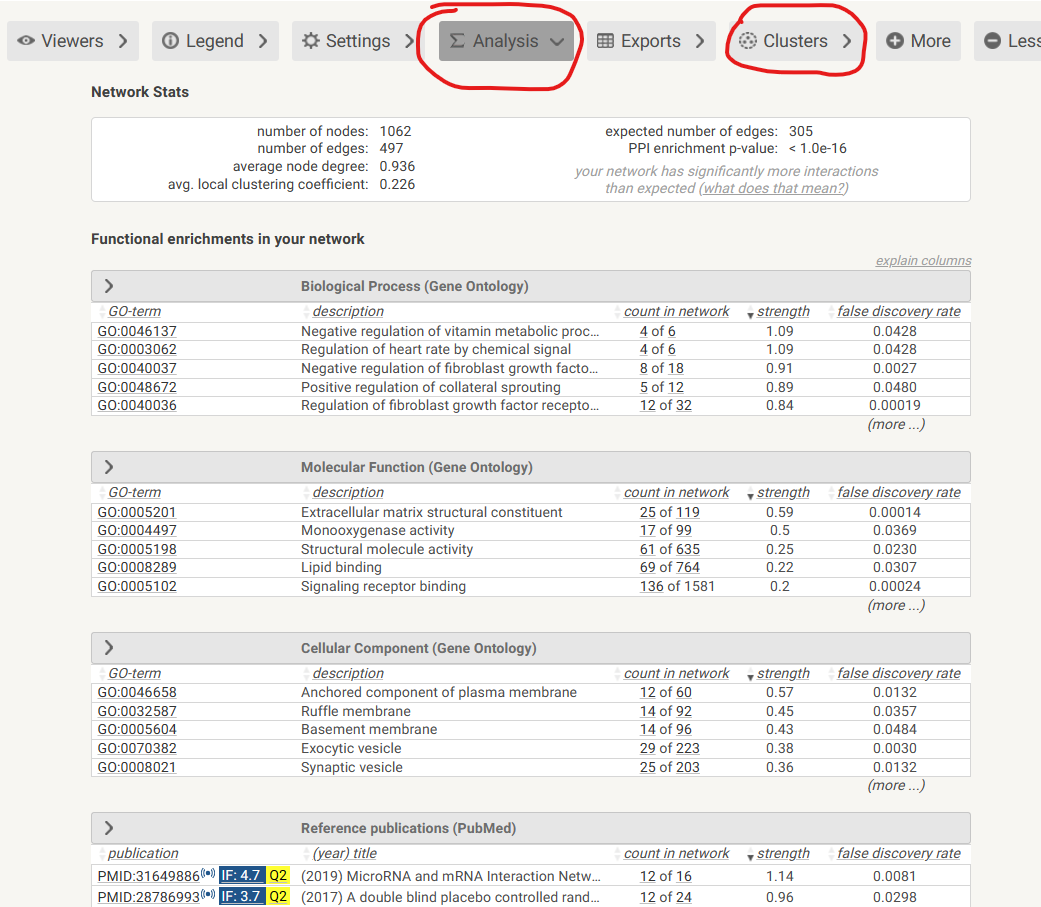
4. 其他分析
主要包括富集分析和聚类分析,大家自行尝试:
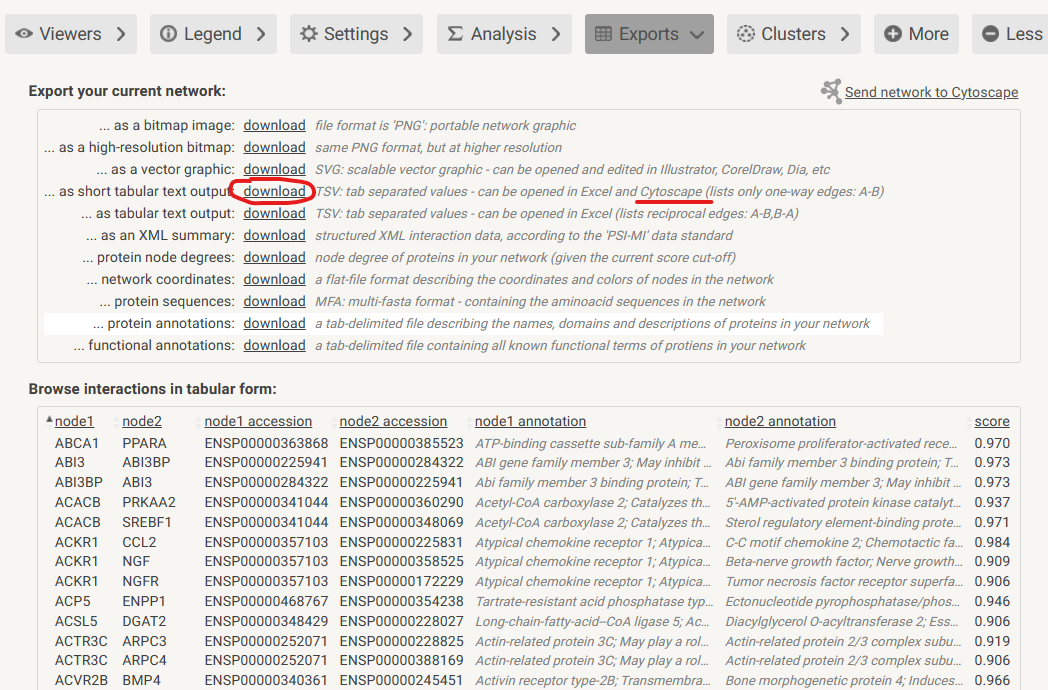
5. 导出tsv数据
用于导入Cytoscape。
6. 导入Cytoscape
第一步是导入数据:File-Import -Network from file,然后选择TSV格式的文件,默认参数🡪OK。得到一个巨丑无比的网络图,看起来也比较复杂。

7. NetworkAnalyzer美化加工
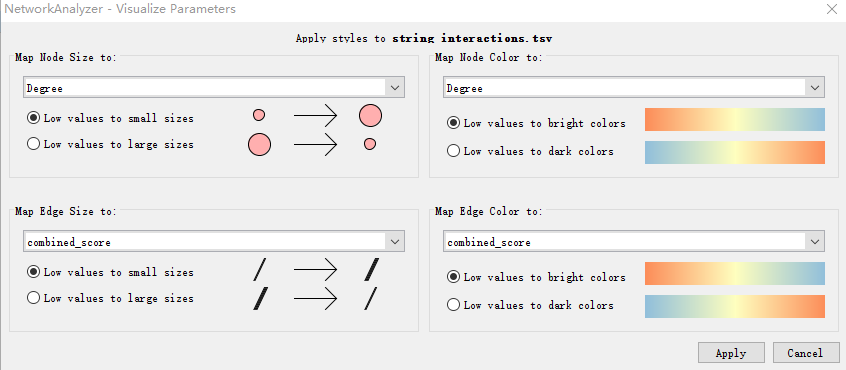
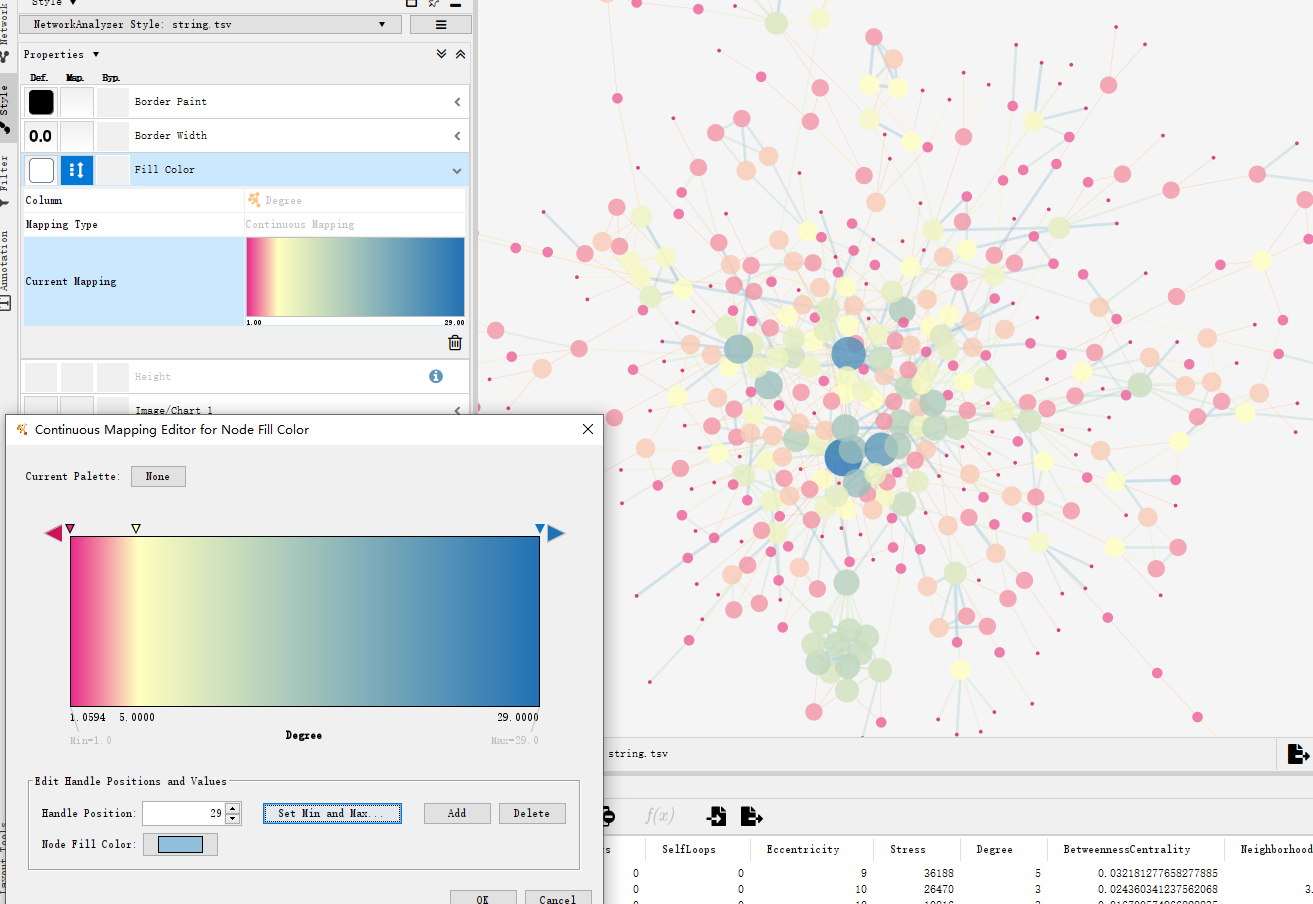
对于Cytoscape V3.7用户可以很方便进行美化:依次点击Tools- NetworkAnalyzer-Network Analysis-Generate style from statistics。
会出现一个弹窗,如图进行参数调整:
本教程使用的Cytoscape V3.9,可能大多数人使用的高版本,Tools下面只有Network Analysis,没有方便大家的Generate style from statistics。Oh NO!!!!!
没有关系,熟悉的朋友应该会手动进行node和edge参数设置,不多说,详见视频教程。
此外,一向贴心的进哥已经给大家制作了一个Style文件,在Cytoscape中import 🡪 Styles from file 🡪 选择NetworkAnalyzer_styles.xml,将预制style导入软件。
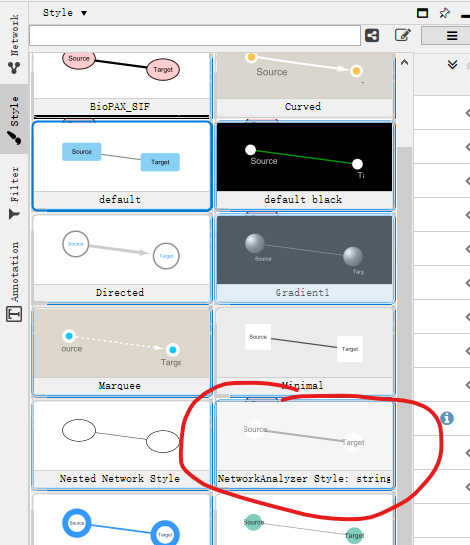
进行Tools🡪Network Analysis后,选择NetworkAnalyzer_styles,瞬间得到和3.7一样的效果:
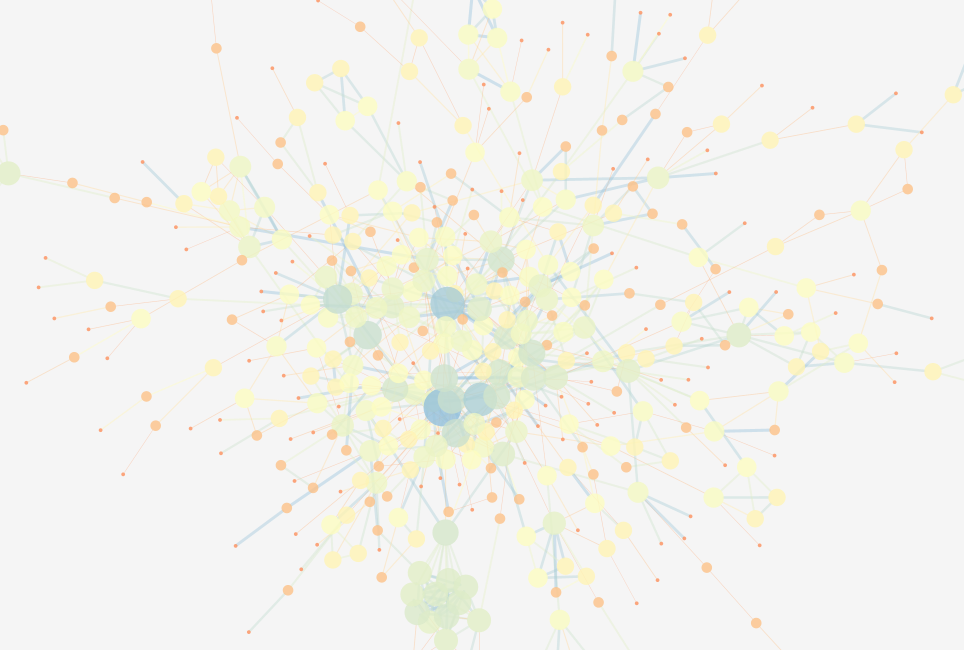
稍加调整颜色参数,使整体更加清晰:
还算OK,其它还有很多调整,按照自己的审美进行微调。
8. 将LogFC信息加入PPI
可能大家大多数看的文献中都是上图呈现效果,其实只要自己熟悉Cytoscape,完全可以将LogFC,甚至P值信息添加进图中。详见进哥B站视频教程。
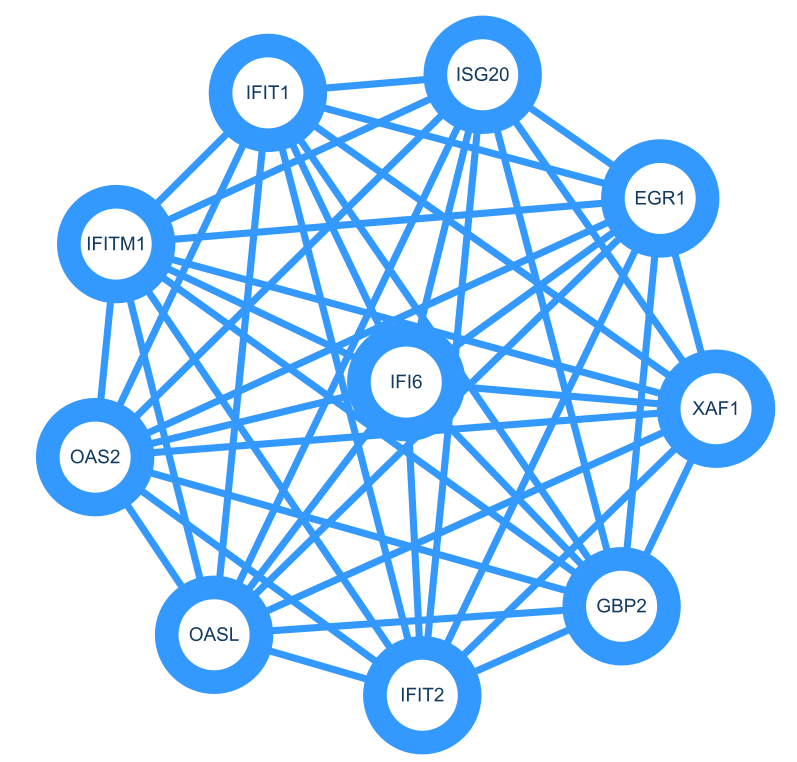
9.寻找核心基因+子网络
一般做完差异基因,或者使用其他方法找到想要的biomarker时,想要知道这些基因的调控网络,或者哪些基因在调控网络中处于核心位置,比较常见的方法就是WGCNA或者MCODE、CYTOHUBBA。今天主要介绍MCODE。
按照上述步骤构建好PPI network之后:Apps🡪 MCODE 🡪 open MCODE🡪 Analyze current network:
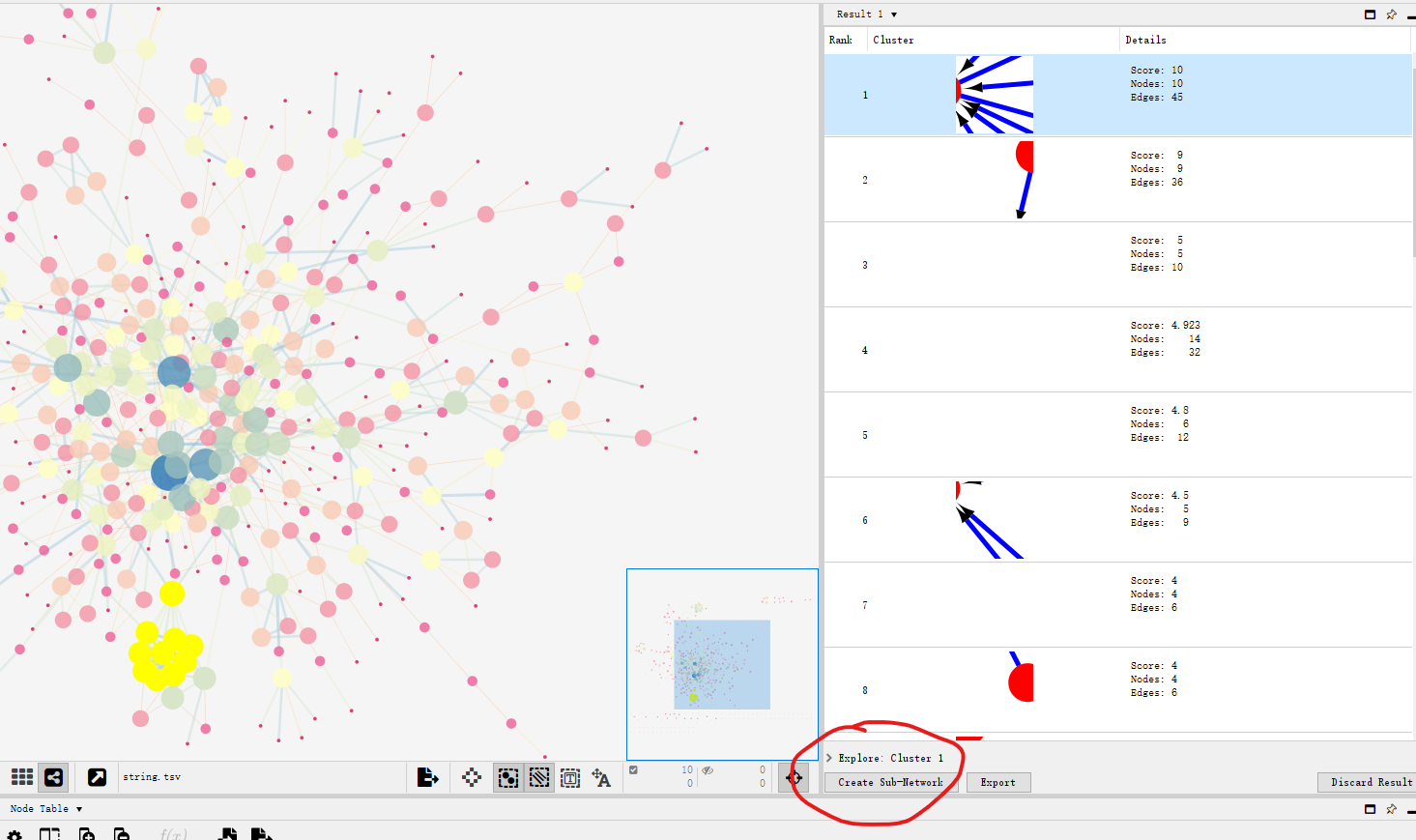
得到多个Clusters,选中其中一个🡪 Create Sub-Network:

由于使用的NetworkAnalyzer_styles,而新的子网络没有相应参数,所以一片苍白,可以再次进行Tools🡪Network Analysis:
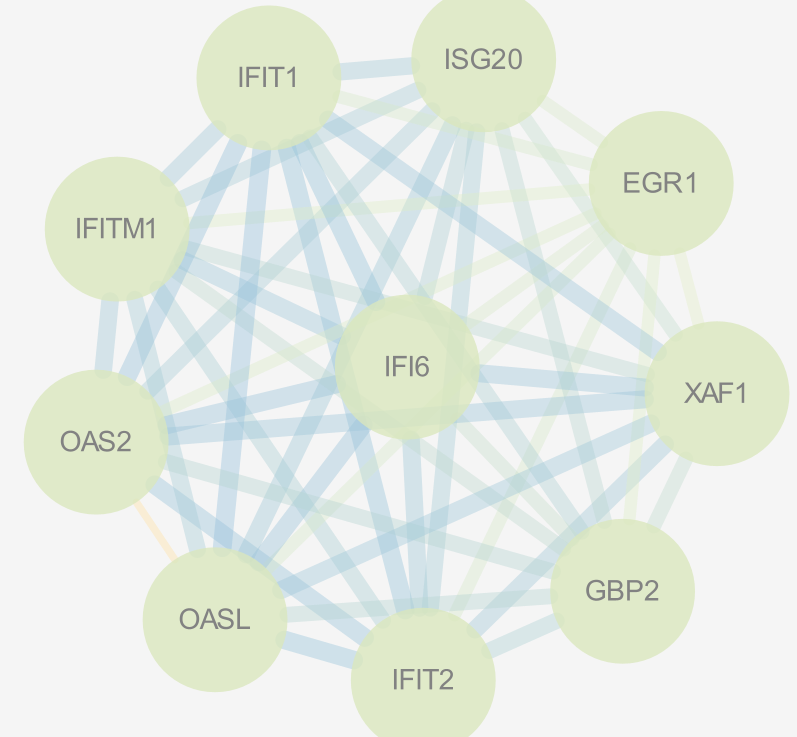
依旧不好看,这个基因便较少,就可以按照自己的喜好自定义子网络,比如换一个style(Ripple):
最后,导出子网络数据:
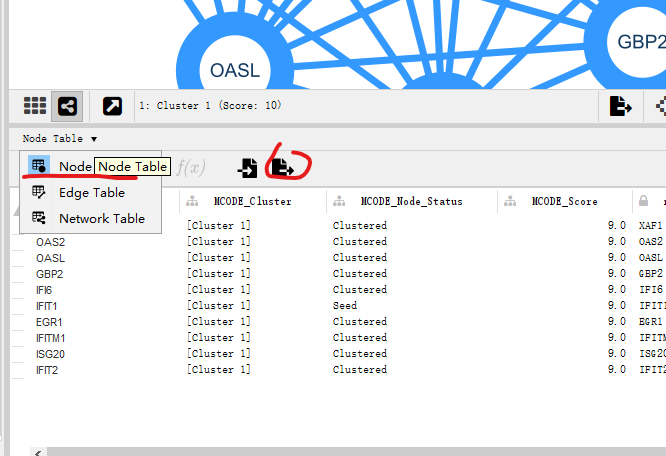
All right!
Cytoscape是一个功能很强大的网络图绘制工具,还有很多扩展插件,大家感兴趣可以自行摸索,会爱上这个工具。
网盘链接:2016-王进邀请您在Filez协作内容: NetworkAnalyzer_styles.xml 点击链接查看: https://console.box.lenovo.com/l/nuNTPt提取码: zclt到期日: 2024-07-18
最后祝大家科研实验顺利!
您好,Mapping gene symbol这一步没有明白为什么是选择第三个。
请问NetworkAnalyzer_styles.xml这个文件在哪里啊?
下载链接过期了哥,还会更新一下不。
进哥,您知道有啥手段或者载体能够实现乳鼠的基因过表达吗(除了构建模式生物)
王老师好!讲解步骤很清晰,我想请问下如何将LogFC加入PPI呀?先谢谢王老师了!
进哥 文末链接到期了QAQ 请问您方便更新下吗?
您好,这个Style文件还可以分享一下吗
进哥想求NetworkAnalyzer_styles.xml 的文件不知道还有没有呢
进哥你好,我想咨询一下转录组测序的结果,在公司云平台做了WGCNA分析,得到的都是一些基因ID代码,有一些在数据库里根本注释不到,这种情况如何通过string网站做ppi分析呢?
进哥,我的连Network Analysis都没有……
进哥!想请教一下,我做出来的差异蛋白也是特别多,上百个,富集分析的结果其实挺明确的,也有意义。但是在做PPI的时候太多了,即使聚类也会有很多个,有的跟富集分析的结果也不太相关,这时候要怎么挑选核心基因呢?是可以展示聚类的多个结果吗?
王博士,cytoscape 3.10.1不能直接通过软件直接下载插件,只能到AppStore网站里下载,现在插件都不显示Download,点击也没反应,总之都没办法下载插件。你那里有MCODE插件文件吗,能否发我一份,急用,谢谢!
王博士,能否发一份MCODE的插件呢,如果有打包的插件集更好,现在官网都下不了啦。
您好,我也是做CRISPR/CAS9敲除的,最近是在做海水模式生物青鳉鱼生殖基因的敲除,F0代明明有测序验证存在非3倍的碱基缺失,但敲除后得到的F1代全都是3倍的碱基缺失,我始终想不明白,于是就用SMART预测了一下蛋白的功能域,然后重新设计了新的2个靶点(这两个靶点分别位于功能域的首尾),F0代测序验证后,全都是3倍的碱基缺失,唯一一个非3倍的碱基缺失,还是因为脱靶(无法使用),现在我想破脑袋也不明白,请问您可以帮我分析一下为什么会出现这种状况吗?感谢!
您好,根据您的描述,我也想不明白,实在不行还是建议双敲吧,把整段基因或者启动子敲掉
进哥 第二步Mapping gene symbol是什么意思呢,可以展开说说吗
就是:很多基因都有别名,而且不止一种别名,所以这一步mapping就是把你input的genelist与String数据库里面的genesymbol进行映射