1.非模式生物GO富集分析
library(AnnotationHub)
hub <- AnnotationHub()
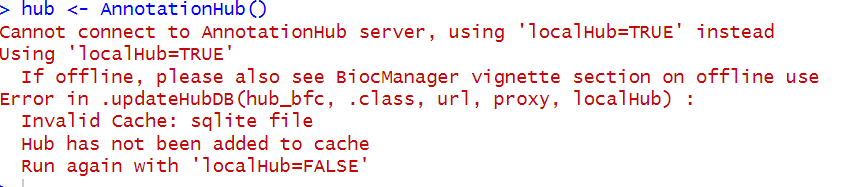
报错了,网上查了好久,终于找到解决方法
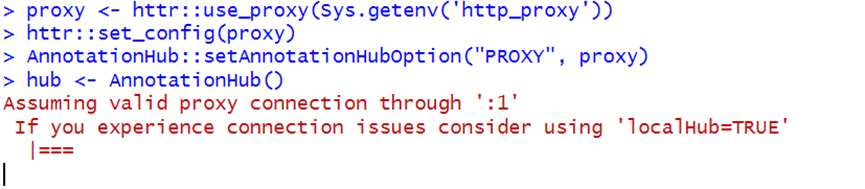
proxy <- httr::use_proxy("http://my_user:my_password@myproxy:8080")
## or
proxy <- httr::use_proxy(Sys.getenv('http_proxy'))
httr::set_config(proxy)
AnnotationHub::setAnnotationHubOption("PROXY", proxy)
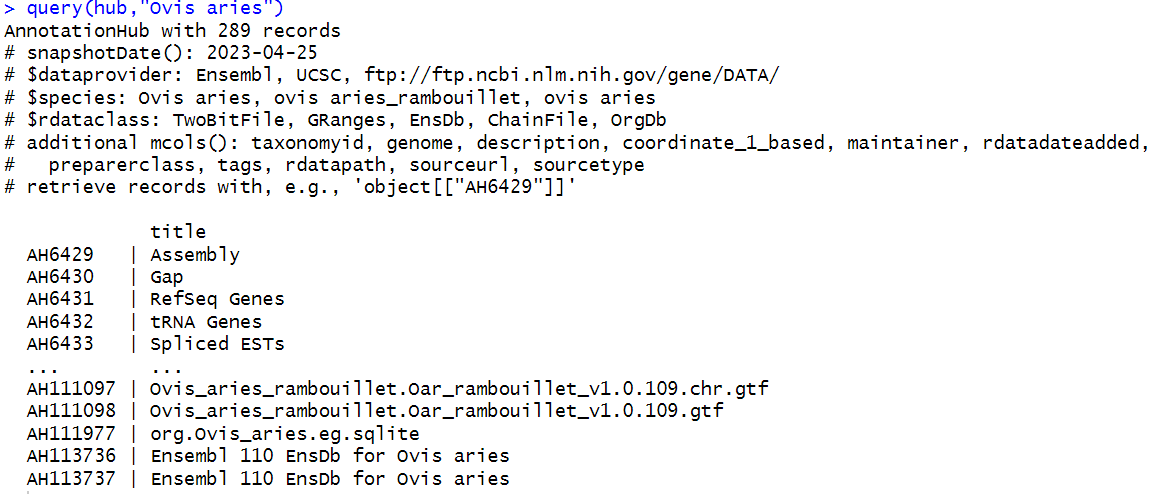
检索绵羊(”Ovis aries”)的数据,此处AH111977是我们所需要的。
# 然后下载这个sqlite数据库
ha.db <- hub[['AH111977']]
#查看前几个基因(Entrez命名)
head(keys(ha.db))
#查看包含的基因数
length(keys(ha.db))
#保存这个数据库,方便后续使用
saveDb(ha.db, "org.oas.eg.sqlite")
#读取数据库
org.oas.eg.db <- loadDb("org.oas.eg.sqlite ")
#接下来可以直接使用org.oas.eg.db进行富集分析
library(org.oas.eg.db)
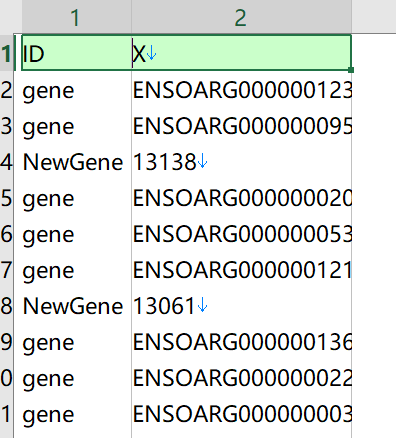
genes= read.delim("result.txt")
require(clusterProfiler)
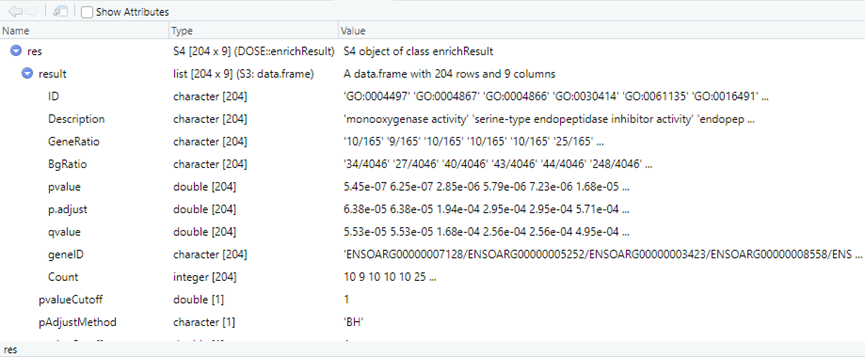
res = enrichGO(genes$X, OrgDb=org.oas.eg.db, keyType = "ENSEMBL",
pvalueCutoff=1, qvalueCutoff=1)
2.将注释文件封装成org.db包(可选)
其实直接loadDB导入.sqlite更方便,此处顺便学习一下,更主要是为了后续加入到之前的富集分析app中。
org.oas.eg.db <- ha.db
library(AnnotationForge)
seed <- new("AnnDbPkgSeed",
Package= "org.oas.eg.db",
Version= "V0.1",
Author= "Jin Wang <jin.wang93@outlook.com>",
Maintainer= "Jin Wang <jin.wang93@outlook.com>",
PkgTemplate="NOSCHEMA.DB",
AnnObjPrefix="org.oas.eg",
organism = "Ovis aries",
species = "Ovis aries",
biocViews = "annotation",
manufacturerUrl = "no manufacturer",
manufacturer = "no manufacturer",
chipName = "no manufacturer")
makeAnnDbPkg(seed, "org.oas.eg.sqlite", dest_dir="org.oas.eg.db")
此时工作目录中会新建一个文件夹org.oas.eg.db,里面是这个包的数据,利用devtools安装这个包。
devtools::install("org.oas.eg.db")
#安装好之后加载这个包。
library(org.oas.eg.db)
res = enrichGO(genes$X, OrgDb=org.oas.eg.db,keyType = "ENSEMBL",
pvalueCutoff=1, qvalueCutoff=1)
3.非模式生物KEGG富集分析
remotes::install_github("YuLab-SMU/createKEGGdb")
createKEGGdb::create_kegg_db('oas')
##此时工作目录下多出一个KEGG.db_1.0.tar.gz,Rstudio中使用Tool-install packages安装该包。
##首先进行ID转化,使用bitr
ttt <- bitr(genes$X, 'ENSEMBL', c("ENTREZID", "GO", "ONTOLOGY"), org.oas.eg.db)
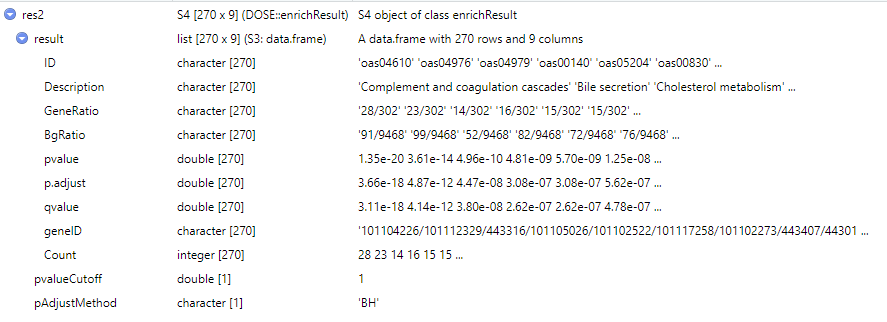
res2 <- enrichKEGG(gene= ttt$ENTREZID,organism= 'oas',
pvalueCutoff = 1,
qvalueCutoff = 1,
use_internal_data =T)
4. 局限
该方法仅限于AnnotationHub中含有注释数据的物种,如果没有,可以使用另一种方法,请参考:构建所需orgDb,用于clusterProfiler富集分析 – 简书