好久没有发布公众号文章和B站视频了,
用户可以直接使用如下链接访问PCAS Shinyapp:https://jingle.shinyapps.io/PCAS/
也可以安装该R包:
remotes::install_github("WangJin93/PCAS") 输入PCAS_app()函数运行PCAS app, 该APP的使用参照本工具的文章:
Citation: Wang J, Song X, Wei M, Qin L, Zhu Q, Wang S, Liang T, Hu W, Zhu X, Li J. PCAS: An Integrated Tool for Multi-Dimensional Cancer Research Utilizing Clinical Proteomic Tumor Analysis Consortium Data. International Journal of Molecular Sciences. 2024; 25(12):6690. https://doi.org/10.3390/ijms25126690IF: 5.6 B2
或B站视频:
https://www.bilibili.com/video/BV1W6421Z7nr/
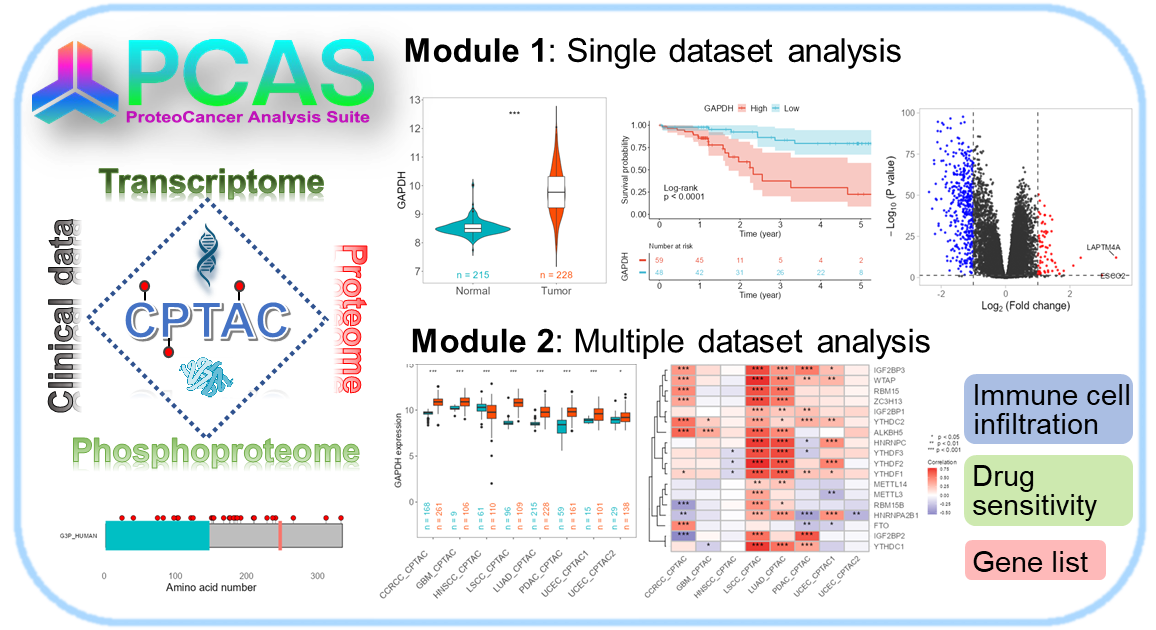
PCAS包主要函数:
- get_data():
Description
Get the CPTAC data by using the api. All results saved in MySQL database.
Usage
get_data(
table = "LUAD_Academia_protein",
action = "expression",
genes = c("GAPDH", "TNS1")
)
Arguments
table | For action = expression, use dataset$Abbre to get all tables; For action = clinic, remove _protein/_mRNA/_Phospho from dataset$Abbre. |
action | “expression”, “degs” or “clinic”. |
gene | Gene symbols, you can input one or multiple symbols. |
2. get_expr_data()
Description
Get the mRNA/protein expression data in CPTAC database.
Usage
get_expr_data(
datasets = c("LUAD_CPTAC_protein", "LSCC_CPTAC_protein"),
genes = c("TP53", "TNS1")
)
Arguments
datasets | Dataset names, you can input one or multiple datasets. Use ‘dataset$Abbre’ to get all datasets. |
genes | Gene symbols, you can input one or multiple symbols. |
3. Get_DEGs_result()
Description
Get the results of different expression analysis between tumor and normal samples in CPTAC datasets.
Usage
get_DEGs_result(dataset = "LUAD_CPTAC_protein", method = "t.test") Arguments
dataset | Use dataset$Abbre to get all tables. |
method | “limma” or “t.test”. |
4. merge_clinic_data()
Description
Get clinic data and merge it with expression data.
Usage
merge_clinic_data(cohort = “LUAD_APOLLO”, data_input)
Arguments
cohort | Data cohort, for example, “LUAD_APOLLO”, “LUAD_CPTAC”. |
data_input | Expression data obtained from get_expr_data() function. |
5. cor_cancer_genelist()
Description
Perform correlation analysis of the mRNA/protein expression data in CPTAC database.
Usage
cor_cancer_genelist(
dataset1 = "LUAD_CPTAC_protein",
id1 = "STAT3",
dataset2 = "LUAD_CPTAC_mRNA",
id2 = c("TNS1", "TP53"),
sample_type = c("Tumor", "Normal"),
cor_method = "pearson"
)
Arguments
dataset1 | Dataset name. Use ‘dataset$Abbre’ to get all datasets. |
id1 | Gene symbol, you can input one gene symbols. |
dataset2 | Dataset name. Use ‘dataset$Abbre’ to get all datasets. |
id2 | Gene symbols, you can input one or multiple symbols. |
sample_type | Sample type used for correlation analysis, default all types: c(“Tumor”, “Normal”). |
cor_method | cor_method for correlation analysis, default “pearson”. |
6. cor_pancancer_genelist()
Description
Perform correlation analysis of the mRNA/protein expression data in CPTAC database.
Usage
cor_pancancer_genelist(
df,
geneset_data,
sample_type = c("Tumor", "Normal"),
cor_method = "pearson"
)
Arguments
df | The expression data of the target gene in multiple datasets, obtained by the get_expr_data() function. |
geneset_data | The expression data of a genelist in multiple datasets, obtained by the get_expr_data() function. |
sample_type | Sample type used for correlation analysis, default all types: c(“Tumor”, “Normal”). |
cor_method | Method for correlation analysis, default “pearson”. |
7. cor_pancancer_drug()
Description
Calculate the correlation between target gene expression and anti-tumor drug sensitivity in multiple datasets.
Usage
cor_pancancer_drug(
df,
cor_method = "pearson",
Target.pathway = c("Cell cycle")
)
Arguments
df | The expression data of the target gene in multiple datasets, obtained by the get_expr_data() function. |
cor_method | Method for correlation analysis, default “pearson”. |
Target.pathway | The signaling pathways of anti-tumor drug targets, default “Cell cycle”. Use “drug_info”to get the detail infomation of these drugs. |
8. cor_pancancer_TIL
Description
Calculate the correlation between target gene expression and immune cells infiltration in multiple datasets.
Usage
cor_pancancer_TIL(df, cor_method = "spearman", TIL_type = c("TIMER")) Arguments
df | The expression data of the target gene in multiple datasets, obtained by the get_expr_data() function. |
cor_method | Method for correlation analysis, default “pearson”. |
TIL_type | Algorithm for calculating immune cell infiltration, default “TIMER”. |
9. viz_TvsN()
Description
Visualizing the different expression of mRNA/protein expression data between Tumor and Normal tissues in CPTAC database.
Usage
viz_TvsN(
df,
df_type = c("single", "multi_gene", "multi_set"),
Show.P.value = TRUE,
Show.P.label = TRUE,
Method = "t.test",
values = c("#00AFBB", "#FC4E07"),
Show.n = TRUE,
Show.n.location = "default"
)
10.viz_DEGs_volcano()
Description
Plotting volcano plot for DEGs between tumor and normal samples in CPTAC datasets.
Usage
viz_DEGs_volcano(
df,
p.cut = 0.05,
logFC.cut = 1,
show.top = FALSE,
show.labels = NULL
)
Arguments
cohort | Data cohort, for example, “LUAD_APOLLO”, “LUAD_CPTAC”. |
data_input | Expression data obtained from get_expr_data() function. |
11. viz_cor_heatmap()
Description
Presenting correlation analysis results using heat maps based on ggplot2.
Usage
viz_cor_heatmap(r, p) Arguments
r | The correlation coefficient matrix r of the correlation analysis results obtained from the functions cor_pancancer_genelist(), cor_pancancer_TIL(), and cor_pancancer_drug(). |
p | The P-value matrix p of the correlation analysis results obtained from the functions cor_pancancer_genelist(), cor_pancancer_TIL(), and cor_pancancer_drug(). |
12. viz_corplot()
Description
Scatter plot with sample size (n), correlation coefficient (r) and p value (p.value).
Usage
viz_corplot(
data,
a,
b,
method = "pearson",
x_lab = " relative expression",
y_lab = " relative expression"
)
Arguments
data | A gene expression dataset with at least two genes included, rows represent samples, and columns represent gene expression in the matrix. |
a | Gene A |
b | Gene B |
method | Method for correlation analysis, “pearson” or “spearman”. |
x_lab | X-axis label. |
y_lab | Y-axis label. |
13. viz_phoso_sites()
Description
Query phosphorylation site information of target proteins based on CPTAC database phosphorylation proteomics data or UniProt database.
Usage
viz_phoso_sites(gene = "YTHDC2", phoso_infoDB = "CPTAC") Arguments
gene | Gene/protein symbol. |
phoso_infoDB | Database for extracting phosphorylation site information. only supports ‘UniProt’ and ‘CPTAC’, Default “CPTAC”. |
14. Citation:
Wang J, Song X, Wei M, Qin L, Zhu Q, Wang S, Liang T, Hu W, Zhu X, Li J. PCAS: An Integrated Tool for Multi-Dimensional Cancer Research Utilizing Clinical Proteomic Tumor Analysis Consortium Data. International Journal of Molecular Sciences. 2024; 25(12):6690. https://doi.org/10.3390/ijms25126690
进哥,您好,我想问一下
1.我想获取完整的蛋白质表达矩阵和对应的mRNA表达矩阵应该怎么做?
2.在得到蛋白质表达矩阵后,想要得到差异表达蛋白您是怎么做的,例如NA值如何处理,,logFC如何计算(我看github上有些是用平均值相减)
3蛋白质表达矩阵应该如何处理才能进行多因素cox和lasso回归