前言
肿瘤组织不是单纯的只包含肿瘤细胞,它是由各种不同类型的细胞组成的,其中就包含基质细胞,成纤维细胞,还有免疫细胞等,这些细胞就构成了肿瘤的微环境。
我们比较关注的是免疫细胞在这微环境中的作用。免疫细胞又包括很多种,如 B 细胞,T 细胞等。不同的免疫细胞在肿瘤发生过程中发挥不同的作用,而不同肿瘤的免疫细胞组成也各有特点。
因此在研究肿瘤发生,治疗等机制时,常常会对不同肿瘤类型的免疫细胞进行定量研究。所谓的定量研究就是研究不同免疫细胞的比例。
为了准确的评估肿瘤微环境中免疫细胞的构成,我们可以通过很多方法从 RNA 测序数据中量化肿瘤浸润的免疫细胞,比如 ssGSEA, Cibersoft, TIMER, EPIC 等.
这些方法大体包括两个思想,一个是 marker gene,还有一个是反卷积思想。
今天我们主要是讲讲如何使用这些方法来进行相关分析。我们通过测序得到的组织的 RNA-Seq 数据就是包含了肿瘤和微环境细胞共同表达。因此在我们进行免疫浸润分析的时候,基本都是基于 RNA-Seq 数据。
1 ssGSEA
单样本基因集富集分析(single sample gene set enrichment analysis, ssGSEA),是针对单个样本无法做 GSEA 而设计的。最早是在 2009 年被提出(Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1)。它可以由 GSVA 这个 R 包来实现。目前 ssGSEA 常被用于评估肿瘤免疫细胞浸润程度。
1.1 R 包安装
### GSVA 安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GSVA")
1.2 数据示例
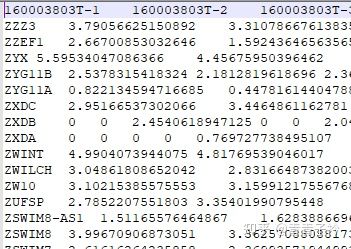
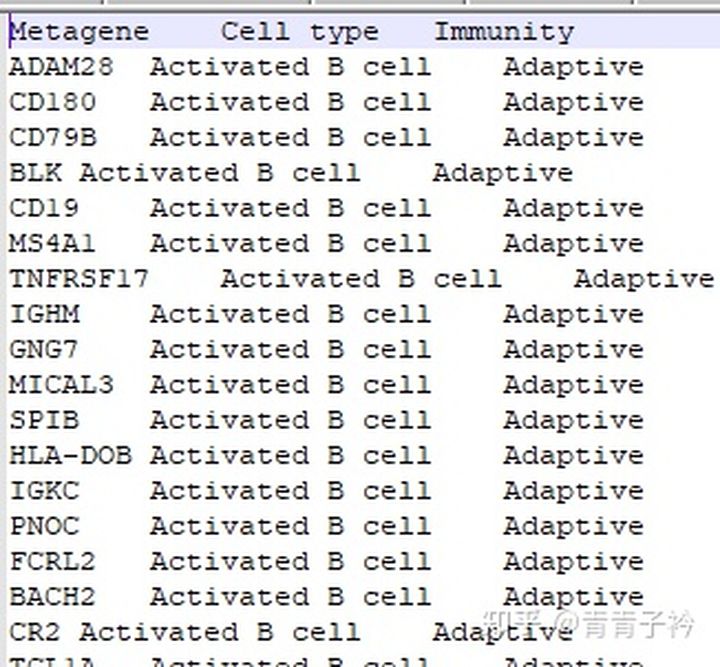
这个数据为免疫细胞的 metagenes 基因集,从文章 Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer 中获取。
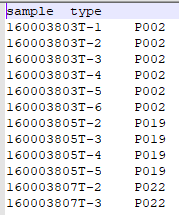
1.3 R 代码实现
library(GSVA)
# 基因表达数据文件
expression <-"mRNA_expression.txt"
# 读取表型数据文件
pheno <- "mRNA_group.txt"
# 背景基因集合文件
gene_set <- "gene_set.txt"
# 读取基因表达数据
expression <- read.table(expression,sep="\t", header=T, row.names = 1)
# 读取背景基因集合
gene_set <- read.table(gene_set,header=T, sep="\t", stringsAsFactors = F)[,c(1:2)]
# 存储的是每个免疫细胞对应的基因,构建背景基因集合
bg_genes <- split(as.matrix(gene_set)[,1], gene_set[,2])
# 进行 ssGSEA 分析
gsva_matrix <- gsva(as.matrix(expression), bg_genes, method='ssgsea', kcdf='Gaussian', abs.ranking=TRUE)
# 输出结果
write.table(gsva_matrix,"gsva_matrix.txt", sep="\t", quote=FALSE, row.names = TRUE)
# 绘制图形
library(pheatmap)
# 样本的类型信息,为了后面画图的时候,加个注释条
pheno <- read.table(pheno,header=T,stringsAsFactors = F)
# 添加注释信息
annotation_col = data.frame(patient=pheno$type)
rownames(annotation_col)<-colnames(gsva_matrix)
# 保存图形结果
pdf(file="pheatmap_ssGSEA.pdf")
pheatmap(gsva_matrix,
show_colnames = F, # 不展示行名
cluster_rows = F, # 不对行聚类
cluster_cols = F, # 不对列聚类
annotation_col = annotation_col, # 加注释
cellwidth=5,cellheight=5, # 设置单元格的宽度和高度
fontsize=5) # 字体大小
dev.off()
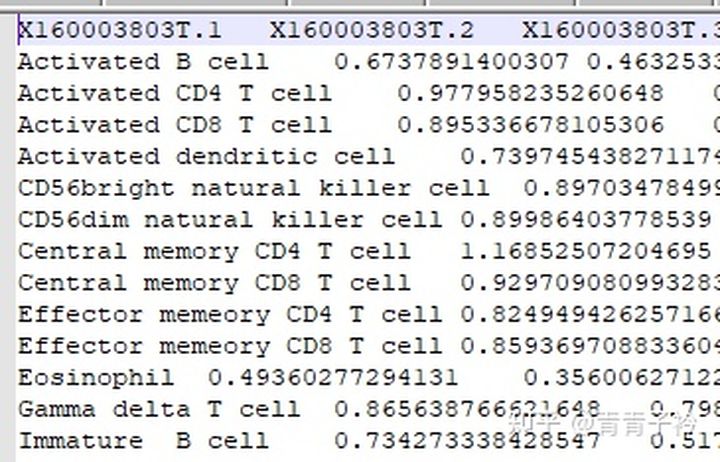
1.4 结果展示
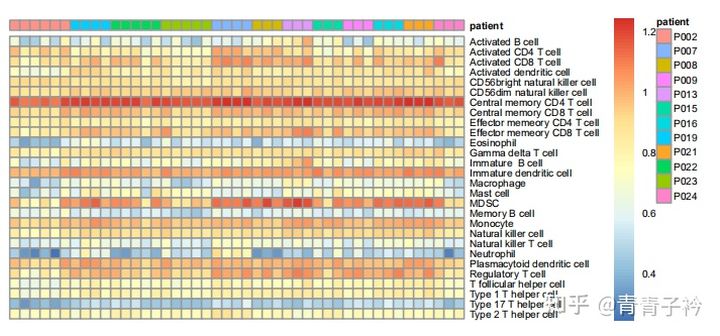
2 其他方法
前面我们说到有很多评估免疫细胞浸润的方法,其中 immunedeconv 就是一个集成很多量化肿瘤浸润的免疫细胞的 R 包。
2.1 immunedeconv 的安装
这个 R 包不同于普通的 R 包,是直接在 GitHub 里,因此它的安装方式有两种,推荐第一种。
使用 conda 安装
conda create -n deconvolution
conda activate deconvolution
conda install -c bioconda -c conda-forge r-immunedeconv
使用 remotes 安装
install.packages("remotes")
remotes::install_github("icbi-lab/immunedeconv")
GitHub 地址
https://github.com/icbi-lab/immunedeconv
2.2 示例
immunedeconv 实现了多种免疫浸润分析方法,包括 quantiseq, timer, cibersort, cibersort_abs, mcp_counter, xcell, epic。
例如
deconvolute(exprMatrix, “quantiseq”)。
下面我们就以 quantiseq 方法来举例说明。
library(ggplot2)
library(immunedeconv)
library(tidyverse)
# 读入表达数据
exprMatrix <- read.table("expression.txt",header=TRUE,row.names=1, as.is=TRUE)
# 使用 quantiseq 方法评估免疫浸润,method 可根据需求设置
res <- deconvolute(exprMatrix, method="quantiseq")
write.table(res, "quantiseq.txt", sep="\t", col.names=T, row.names=F, quote=F)
save(res, "quantiseq.Rdata")
# 画图
pdf("quantiseq.pdf")
res %>%
gather(sample, fraction, -cell_type) %>%
# 绘制堆积条形图
ggplot(aes(x=sample, y=fraction, fill=cell_type)) +
geom_bar(stat='identity') +
coord_flip() +
scale_fill_brewer(palette="Paired") +
scale_x_discrete(limits = rev(levels(res)))
dev.off()
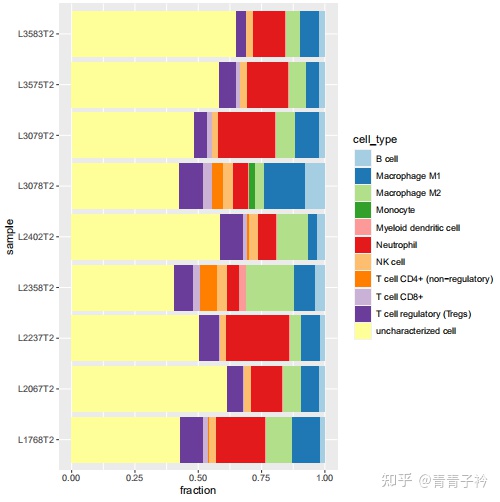
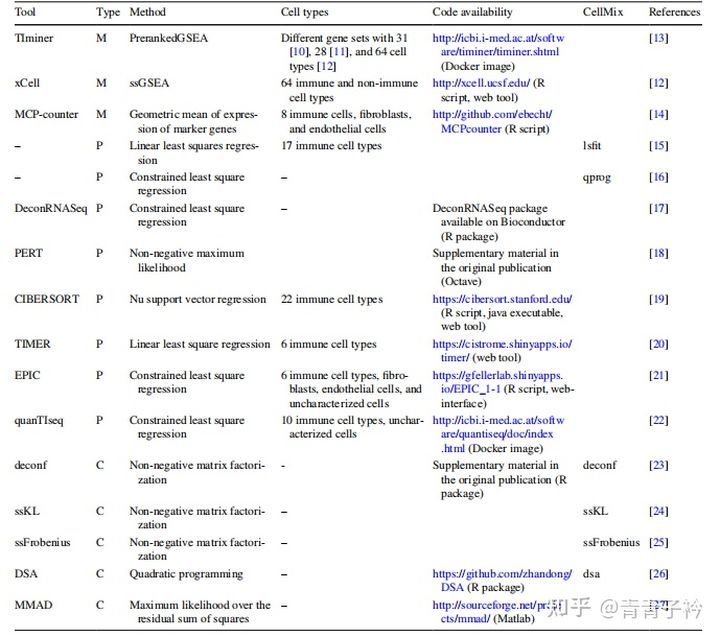
3 分析方法的简单介绍
最后,咱们简单的聊下几个常用的免疫细胞浸润分析方法。
3.1 quanTIseq
quanTIseq 是用于根据人类 RNA-seq 数据量化肿瘤免疫状况,通过反卷积量化样本中存在的十种不同免疫细胞类型的比例以及其他未表征细胞的比例。
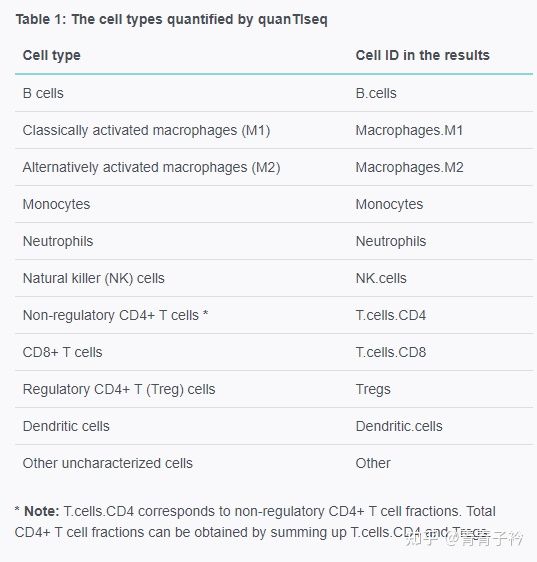
链接:https://icbi.i-med.ac.at/software/quantiseq/doc/index.html
3.2 xCell
xCell 是基于 ssGSEA 的方法,可根据 64 种免疫细胞和基质细胞类型的基因表达数据进行细胞类型富集分析。xCell 使用表达水平排名而不是实际值,因此归一化没有影响,但是输入数据需要 normalization 格式(RPKM/FPKM/TPM/RSEM)。
可使用官网上传的表达数据,行是基因名,列是样本。
3.3 TIMER
TIMER 用反卷积方法估算 32 种癌症中 6 种免疫细胞的丰度(B 细胞,CD4+ T 细胞,CD8+ T 细胞,嗜中性粒细胞,巨噬细胞和树突状细胞)。
TIMER 有在线的网站,功能强大,通过该软件对 TCGA 中的肿瘤样本的表达谱数据进行分析,将肿瘤浸润的免疫细胞与基因的表达量,基因突变,体细胞拷贝数变异等数据相关联。
网址:https://cistrome.shinyapps.io/timer/
3.4 EPIC
EPIC 使用约束最小二乘回归将非负性约束条件纳入反卷积问题,从大量肿瘤基因表达数据中估算免疫和癌细胞的比例,每个样本中所有细胞分数的总和不超过一。
链接:https://gfellerlab.shinyapps.io/EPIC_1-1/
3.5 CIBERSORT
CIBERSORT 是非常常用的一个计算免疫细胞浸润的方法,它利用线性支持向量回归的原理对免疫细胞亚型的表达矩阵进行去卷积,来估计免疫细胞的丰度。
CIBERSORT 提供了 22 种常见的免疫浸润细胞表达数据 LM22,包括不同的细胞类型和功能状态的免疫细胞。
CIBERSORT 有在线的网站,但是需要注册教育邮箱账号,操作也十分简单,只要用户上传表达数据即可
官网链接:https://cibersort.stanford.edu/。
具体操作可参考:https://www.jianshu.com/p/a8759484359c。
CIBERSORT 还开发了可适用于单细胞测序的方法–CIBERSORTx。这是一种机器学习方法,可将此框架扩展为无需物理细胞分离即可推断特定细胞类型的基因表达谱。
链接:https://cibersortx.stanford.edu/
3.5.1 R 代码实现
其中 LM22.txt,Cibersort.R 都可以从官网获得,LM22.txt 是 22 种常见的免疫浸润细胞表达数据。
# 可以从官网下载
source('Cibersort.R')
# 其中 nperm 指定的是置换的次数,QN 分位数归一化,如果是芯片设置为 T,如果是测序就设置为 F,测序数据最好是TPM
res_cibersort <- CIBERSORT('LM22.txt','Data.txt', perm = 10, QN = F)
write.table(res_cibersort, file="res_cibersort.txt", sep="\t", col.names=T, row.names=F, quote=F)
save(res_cibersort, file="res_cibersort.Rdata")
# 画图
library(ggplot2)
library(tidyverse)
colour <- c("#DC143C","#0000FF","#20B2AA","#FFA500","#9370DB","#98FB98","#F08080","#1E90FF","#7CFC00","#FFFF00","#808000","#FF00FF","#FA8072","#7B68EE","#9400D3","#800080","#A0522D","#D2B48C","#D2691E","#87CEEB","#40E0D0","#5F9EA0","#FF1493","#0000CD","#008B8B","#FFE4B5","#8A2BE2","#228B22","#E9967A","#4682B4","#32CD32","#F0E68C","#FFFFE0","#EE82EE","#FF6347","#6A5ACD","#9932CC","#8B008B","#8B4513","#DEB887")
my_theme <- function(){
theme(panel.grid = element_blank(), # 网格线
panel.border = element_blank(), # 面板边框
legend.position="right", # legend位置
legend.text = element_text(size=8), # legend内容大小
legend.title = element_text(size=8),# legend标题大小
axis.line = element_line(size=1), # 坐标轴线
text = element_text(family="Times"),# 文本字体
axis.text.y = element_text(size = 8,face='bold',color='black'),# y轴标签样式
axis.text.x = element_text(size = 8,face='bold',color='black',angle=90,hjust=1), # x轴标签样式,angle=45 倾斜 45 度
axis.title = element_text(size=10,face="bold"), # 轴标题
plot.title = element_text(hjust=0.5,size=10)) # 距,设置绘图区域距离边的据类,上、右、下、左
}
p1 <- res_cibersort[,1:22] %>% reshape2::melt() %>%
ggplot(aes(x=Var1,y=value,fill=Var2)) +
geom_bar(stat='identity') +
coord_flip() +
scale_fill_manual(values =colour ) +
theme_bw()+ theme(panel.border = element_blank(),panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),axis.line = element_line(colour = "black"))+my_theme()
pdf("cibersort.pdf")
p1
dev.off()
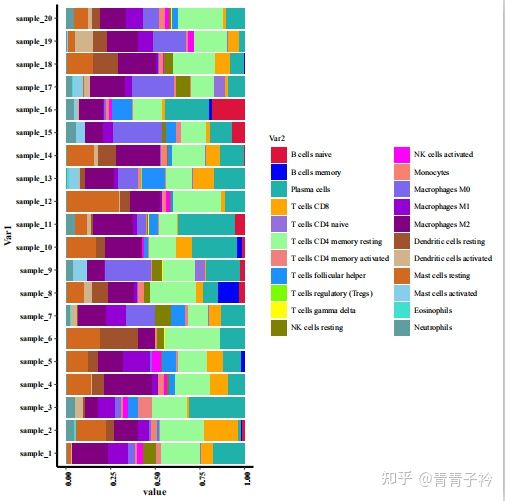
好啦,免疫浸润的方法暂时就介绍到这了,具体的更深入的不同免疫浸润方法的比较可以参考文献
Quantifying tumor-infiltrating immune cells from transcriptomics data
上述代码已经上传到 GitHub:
https://github.com/dxsbiocc/learn/tree/main/R/immune
本文转自:生信学习手册
您好,请问哪些方法适合大鼠样本呢,我了解的xcell只可以做人的。感谢~
王老师,GSVA更新了,做ssgsea有一些变动
大神,请问是否可以用xCell或者是cibersort的免疫细标志物基因集带入到ssGSEA算法去估计免疫浸润?
可以教一下lncrna该怎么做免疫浸润分析吗
和mRNA一样的分析 有各个样本的免疫浸润评分和目的基因表达量,进行相关性分析 明白吗?
想请问一下immunedeconv包中mcp_count算法
mcp_counter <- deconvolute(input, method="mcp_counter")会报错:
“Error in open.connection(file, "rt") :
Could not resolve host: raw.githubusercontent.com”
我看immunedeconv帮助中没有相关解释
您好 其实就是打不开这个链接 网络原因 国内访问不稳定
老师好!想问一下, 目前ssGSEA 常被用于评估肿瘤免疫细胞浸润程度。能否用来其他功用?比如用ssGSEA计算数据库中某疾病的铜死亡得分,再比较每个基因表达值与ssGSEA得分的相关性,筛选铜死亡相关基因?
当然可以的,这只是一个算法,根据目的选用不同的基因集
王老师,您好,请问用CIBERSORT计算免疫细胞的比例时,发现有三种免疫细胞在每个样品中比例全为零,在这种情况下,得到的结果还能用吗?
您好 这个具体我也不清楚 您表达矩阵没有取对数吧,这个要求是不取log的数据,如果用的log,那转换为原始数据分析看看,而且数据不能有缺失,如果有 用knn算法补足。如果还是这样,就使用分析的结果好了
王老师,您好。免疫浸润的新手一枚,请教个问题,使用转录组RNA-seq数据进行免疫浸润分析,看每种组织中不同免疫细胞的比例,即默认每种组织中不同免疫细胞的比例之和均为1,那么不同种组织中免疫细胞的总和是否相同?是否可以从RNA-seq数据中看出每种免疫细胞的“绝对值”,而不只是相对值?
您好,这个算出来的是每种细胞的比例,不管什么组织,总和都是1
绝对值是得不到的,也可能是我积累有限,如果有相关文献,欢迎一起讨论
王老师,有resident macrophages相关的分析吗
您好 这个我没有诶,你如果有相关资料可以分享一下 我看到也发你
王老师,您好,麻烦问下immunedeconv包和cibersort.R这些包做免疫浸润分析时需要的表达矩阵要最原始的没有log处理过的,还是经过log处理过的标准化数据?
按照包说明文档是这样的The input data is a gene ×× sample gene expression matrix. In general values should be
TPM-normalized
not log-transformed.
不取对数的tpm数据
其它的可以试试,问题应该也不大
嗯,网上两个都有,可以选择转换成人源后进行分析,也可以不转换直接使用其他数据库进行分析。只是当时自己做的时候因为CIBERSORT是使用人源的所以一直在寻求一个好的转换方法,后来也解决了重复问题。谢谢老师
conda安装immunedeconv也有报错
不清楚诶。conda不是Python环境管理工具吗?这个immunedeconv是R语言包,也可以?
包的问题解决啦。现在的问题是,手上一批小鼠RNA-seq数据,在基因转换成人的时候,总是出现重复,不知道如何取舍,王老师能给点建议么?谢谢
您好,刚刚看到您的回复。为什么要把小鼠的转化成人的,没有对应的小鼠免疫浸润的资料?我不清楚这样转换是否合适,如果您有相关参考资料,不放留言,我也学习一下。但是就重复这个情况可以参考这个里面的函数,无非取平均值中位数最大值等等GEO数据下载及提取表达矩阵,除去这个 您还可以将鼠的基因名字与您转换后的用“_”连接起来作为新的名称也未尝不可,这样不会涉及数据缺失
大神,求MDSC的TIDE算法
您好,作者好像没有给出算法的代码,不过可以在线提交,结果几秒就出。官网有教程,这边也整理了一下:TIDE算法来预测免疫治疗疗效